

Abstract
Treatment options for patients with sickle cell disease (SCD) continue to rapidly expand and evolve. The goal of therapies such as an allogeneic hematopoietic stem cell transplant (HSCT), gene therapy, and gene editing is to cure rather than control SCD. The benefits of these therapies must be accompanied by minimizing long-term adverse health outcomes from SCD and its treatment. SCD can have adverse effects on a variety of organ systems, including the heart, lung, kidney, and reproductive system, leading to high disease burden, morbidity, and premature mortality in both pediatric and adult patients. While curative therapies are being increasingly used, there remains a paucity of data on the long-term health outcomes associated with these treatments in children and adults with SCD. There are data available regarding the effects of HSCT performed largely for malignant diseases, from which data on SCD outcomes may be extrapolated. However, given the significant differences between these 2 populations of patients who undergo HSCT, such extrapolation is imprecise at best. Furthermore, there are currently no published data on long-term health outcomes following gene therapy for SCD due to current short follow-up times. We summarize the limited data reported on health outcomes following HSCT for SCD and emphasize the need for more research within this area.
Learning Objectives
Describe the various curative therapies available for patients with sickle cell disease
Summarize the current literature on late effects of an allogeneic hematopoietic stem cell transplant for sickle cell disease
Understand the need for continued research on long-term health outcomes of curative treatments for sickle cell disease
CLINICAL CASE
A 15-year-old boy with sickle cell disease (SCD) presents with his parents to your stem cell transplant clinic as a referral from his primary hematologist to discuss curative therapy for his SCD. Despite initiating treatment with hydroxyurea at a young age, he has experienced numerous disease-related complications. He requires chronic transfusions due to a history of cerebral infarctions and has had numerous admissions for acute chest syndrome, vaso-occlusive episodes requiring chronic opioids and several hospitalizations, moderate persistent asthma, and delayed puberty.
You perform a complete evaluation, including physical examination, laboratory tests, and imaging. The patient is found to have severe organ toxicity related to his SCD, including proteinuria, with an echocardiogram revealing an elevated tricuspid regurgitant jet velocity (TRJV) of 2.7 m/s and obstructive lung disease with pulmonary function tests (PFTs) showing a predicted percent forced expiratory volume in 1 second (FEV1) of 65%. You determine that he is a candidate for curative therapy and discuss various treatment options.
Introduction
SCD is a severe inherited disorder of red blood cells resulting from a single nucleotide change in the β-globin gene leading to rigid red blood cells with impaired oxygen-carrying capacity.1 Approximately 100 000 individuals in the United States are affected by SCD, and about 1 in 365 children born of African descent is diagnosed with SCD.2 Although first described in the literature in 1910, the pathophysiology of SCD was not understood until several decades later. Early detection, medications such as hydroxyurea, and a multidisciplinary approach to care have revolutionized the management of SCD. Over the past several years, new agents such as L-glutamine, voxelotor, and crizanlizumab have allowed for expanded options for disease control (Figure 1). Despite this, individuals with SCD can experience a variety of complications, including pain; cardiovascular, pulmonary, and renal disease; stroke; cognitive defects; and reproductive dysfunction. While mortality in children with SCD has decreased over the years,3 adults, particularly those with organ dysfunction, continue to have decreased survival rates.4 Furthermore, the high burden of this disease leads to decreased quality of life and negative psychosocial impacts in children and adults.5
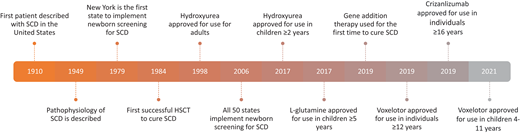
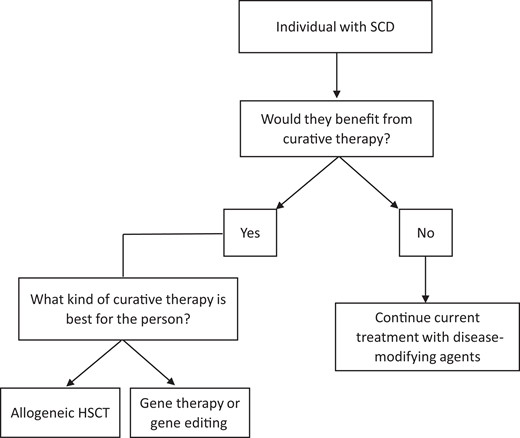
Allogeneic hematopoietic stem cell transplant (HSCT), gene therapy, and gene editing are also now available and the only proven curative options for SCD. HSCT, first successfully performed for SCD in 1984,6 replaces the stem cells in a patient with healthy donor stem cells that produce normal hemoglobin. Gene therapy, first performed in a patient with SCD in 2017,7 introduces a gene that replaces sickled hemoglobin with normal hemoglobin. There are 4 types of gene therapy, including gene addition, gene editing, gene silencing, and gene correction.8 Clinical trials have been essential in continuing to advance the safety and efficacy of gene therapy and editing.9 Determining the appropriate treatment option for a patient is a complex decision and highly individualized. Certain disease-related complications may make curative therapy more advantageous (Figure 2). While these therapies have been successful in curing patients, they are accompanied by risks and long-term effects. There are some data on late effects following HSCT; however, most are in relation to transplant for malignant diseases and extrapolated to SCD. At this time, given the recent success of gene therapy and limited patients with minimal follow-up time, there are no published data on long-term health outcomes for gene therapy, so we present only the data following allogeneic HSCT. There is a critical need for more specific data on long-term outcomes following all curative therapies specifically for SCD given that these therapy options are being more commonly employed.
CLINICAL CASE (Continued)
After discussing various options, you recommend an allogeneic HSCT. Human leukocyte antigen (HLA) typing is performed on all family members, and an unrelated donor search is also initiated. The patient's mother is found to be a haploidentical match and deemed the best suitable donor. The patient and family ask you about the long-term effects of HSCT. Below we summarize the data available.
Cardiovascular disease
An elevated TRJV, pulmonary and systemic hypertension, and systolic and diastolic dysfunction are well-established cardiac complications of SCD. In this section, we focus on the potential long-term implications of HSCT on cardiac function in pediatric and adult patients. In a multicenter prospective study of 19 pediatric patients aged 2 to 20 years who received myeloablative conditioning prior to a haploidentical HSCT, all patients had stable left ventricular shortening fraction (SF) at a median of 2 years posttransplant.10 However, in a larger multicenter retrospective study evaluating 174 pediatric patients who received most commonly a myeloablative conditioning regimen prior to HSCT, 5 patients had worsening ejection fraction and 4 had worsening SF at approximately 4 years post-HSCT.11 A study of 16 patients who received either a myeloablative or reduced intensity conditioning regimen with a median follow-up time of 8.6 years showed a significant decrease in the SF for all patients combined from 41% to 37.5%. However, when evaluating the 3 patients who received a reduced intensity conditioning regimen, there was no change in the SF.12 Adults who undergo a nonmyeloablative conditioning regimen may have improvement in their heart function. In a single-center study of 12 individuals aged 16 to 60 years who received a nonmyeloablative HLA-matched sibling HSCT, patients had a decrease in left atrial diameter posttransplant at 1-year follow-up.13 A slightly larger study of 44 adults who received a nonmyeloablative conditioning prior to a matched sibling or haploidentical HSCT showed improvements in cardiac size, function, and diastolic filling parameters through 1-year follow-up.14
These data suggest that the effects of HSCT on cardiac function vary across study populations, but these data are based on limited sample sizes and variable follow-up times. Overall, more prospective multicenter studies in both pediatric and adult patients are necessary to better understand cardiovascular trajectory post-HSCT.
Pulmonary disease
Pulmonary function impairment is a known complication of SCD. Overall, the limited literature suggests that while FEV1 is a marker of early mortality in adults, the impact on mortality has not been confirmed in children.15 In a multicenter retrospective study of 174 pediatric patients who received mostly myeloablative conditioning prior to HSCT, regardless of whether patients did or did not have lung disease prior to transplant, there was no significant change in pulmonary function posttransplant at a median of 3.2 years' follow-up time.11 Similarly, in a multicenter study of 7 pediatric patients aged 6 to 18 years who received a reduced intensity conditioning regimen, there was no significant change in lung function pre- and posttransplant with variable follow-up time.16 In the largest study of 355 pediatric patients who most commonly received a myeloablative conditioning regimen with an unrelated donor transplant, there was a 2% incidence of pulmonary abnormalities at a median of 4.2 years following transplant.17 Adults may have some improvement in pulmonary function post-HSCT, with 1 study of 12 patients showing improvement in FEV1 and forced vital capacity from baseline after nonmyeloablative conditioning at 1-year follow-up.13 A larger study of 122 adult patients showed stable PFTs with a decrease in the overall proportion of severe lung disease at ~4-year follow-up.18
It is important to understand that the development of graft-vs-host disease (GVHD), particularly chronic GVHD, can be associated with other posttransplant lung disease and can complicate the understanding of pulmonary abnormalities in HSCT survivors.
Overall, the limited literature suggests that while FEV1 is a marker of early mortality in adults, it does not yet appear to be applicable in children. Very limited data suggest that FEV1 may remain stable in children receiving reduced intensity or myeloablative conditioning,16 and adults may see some improvement. More prospective multicenter studies in both children and adults are necessary to determine the course of pulmonary function following curative therapies.
Renal disease
Chronic kidney disease (CKD) is common in adults with SCD and is a known risk factor for early mortality.19 However, little is known about the effects of curative therapies on renal function. One study of 355 pediatric patients treated with mostly myeloablative conditioning regimens prior to HSCT for SCD demonstrated 7 of 355 patients developed sickle cell nephropathy at a median follow-up time of 4.2 years.17 Additional studies have shown that pediatric patients who receive a reduced intensity conditioning regimen may have stable or slightly improved renal function post-HSCT.16,20 There is also some evidence to suggest that hyperfiltration, often an early sign of renal dysfunction in children, may decrease following a nonmyeloablative transplant.21 In a single-center study of pediatric patients who underwent HSCT, 36 of whom for a hemoglobinopathy, 17.1% of patients developed CKD and emphasized the need for close monitoring of renal function.22 In an adult cohort of patients who underwent either a myeloablative or nonmyeloablative transplant, the baseline incidence of CKD prior to transplant was 6% and ranged from 1% to 11% posttransplant.23 This suggests that HSCT in patients with SCD may not increase the risk of renal disease.
Prospectively collected multicenter data on renal function in both pediatric and adult patients remain a gap in current literature.
Reproductive function
Reproductive dysfunction is a well-established complication of SCD; however, HSCT can also lead to complications related to reproductive health, particularly following a myeloablative conditioning regimen.24 Despite advancements in stem cell transplant therapeutic approaches, conditioning regimens contain gonadotoxic medications that lead to an increased risk of impaired fertility. A multicenter trial of 14 pediatric females who underwent an HLA-matched HSCT for SCD revealed that 8 of 14 had evidence of premature ovarian insufficiency and only 4 of 14 had normal estrogen levels. However, 2 females did have successful pregnancies 13 and 14 years following HSCT, respectively. Thirteen pediatric males were also evaluated, with 4 of 13 having low luteinizing hormone/follicle-stimulating hormone levels and 10 of 13 having low testosterone levels.25
All patients should be counseled on the risk of impaired fertility prior to undergoing HSCT. Fertility preservation options such as sperm banking and oocyte/embryo cryopreservation are available for pubertal males and women. Options for prepubertal boys and girls are much more challenging. Certain centers offer ovarian and testicular tissue preservation, some of which remains experimental.26
Once again, dedicated research to understanding fertility following curative therapies for SCD is necessary in order to be able to guide treatment and counsel patients.
CLINICAL CASE (Continued)
The patient undergoes a haploidentical HSCT with a reduced intensity conditioning regimen with thymoglobulin, fludarabine, cyclophosphamide, thiotepa, and 200 cGy total body irradiation. GVHD prophylaxis includes posttransplant cyclophosphamide, sirolimus, and mycophenolate mofetil. Sperm banking was offered; however, the family declined given lack of insurance assistance. His course is complicated by bacteremia but engrafts appropriately and is discharged on day +35. One-year PFTs show stable FEV1 of 66%, and echocardiogram reveals a decrease in TRJV to 2.5 m/s with normal cardiac function (ejection fraction/fractional shortening). He continues to have proteinuria but stable creatine clearance.
Surveillance
There remains a lack of consensus on long-term follow-up surveillance guidelines following HSCT for SCD. The Children's Oncology Group, National Marrow Donor Program, and expert opinion have suggestions for monitoring following HSCT that may include a small proportion of patients with SCD27-29 (Table 1). The Second Pediatric Blood and Marrow Transplant International Consensus Conference on Late Effects after Pediatric HCT provided suggestions specific for hemoglobinopathies including SCD and thalassemia30 (Table 1). In 2022, Fitzhugh et al31 extrapolated data on surveillance following HSCT for hematologic malignancies to provide suggestions for guidelines following HSCT for SCD. Importantly, all of these guidelines are based on limited data, and there are currently no surveillance guidelines following gene therapy. Despite limited evidence, patients are encouraged to receive follow-up care by a team experienced in HSCT survivorship and SCD. Furthermore, relevant subspecialists should be involved in the care of these individuals as necessary given their complexities. It is imperative to dedicate research efforts to long-term health outcomes to allow for more evidence-based, consensus guidelines.
Various surveillance guidelines following hematopoietic stem cell transplant
| Organ system | Late effects | Children's Oncology Group27 | National Marrow Donor Program28 | Bhatia et al29 | SCD specific consortium30 | Fitzhugh et al31 |
|---|---|---|---|---|---|---|
| Cardiovascular | Elevated TRJV, pulmonary and systemic HTN, systolic/diastolic dysfunction, dyslipidemia, cardiac iron overload, myocardial strain | Yearly BP; lipids every 2 years; echo every 1-5 years | Regular monitoring of risk factors and complications but no specific time frame | Yearly BP; lipids every 2 years; echo every 1-5 years | Yearly BP and echo; lipids every 5 years | Yearly BP and echo; lipids every 1-2 years; cardiac MRI monitoring in those with cardiac iron overload before HSCT; cardiology/pulmonology consult for those with severe cardiac disease |
| Pulmonary | Decreased pulmonary function | PFTs at 1-year post-HSCT | PFTs at 6 mo, 12 mo, yearly | PFTs at 1-year post- HSCT | PFTs at 3, 6, and 12 mo and then yearly for 2 years; yearly PFTs for those with early compromise or chronic GVHD until off immunosuppressive medications; echo with evaluation of TRJV at 1 year; pulmonary arterial pressure if TRJV >3 m/s to confirm pulmonary HTN | PFTs at 3, 6, and 12 mo post-HSCT, then yearly; less frequent studies for patients who are not symptomatic, are free of GVHD, and have stable PFTs; pulmonary consultation for those with new or worsening abnormal PFTs |
| Renal | Hyperfiltration, proteinuria, acute/chronic renal disease, sickle cell nephropathy | Renal function for 1 year; yearly UA and BP | BP and renal function tests at 6 mo, 1 year, yearly | Renal function at 1 year; yearly UA and BP | Monitor until off nephrotoxic therapy and yearly for 2 years; UA for blood/protein at 1 year. Microalbuminuria testing yearly for 2 years; yearly BP | Monitor renal function until off nephrotoxic therapy and then yearly; less frequent testing for patients with stable electrolytes and renal function; yearly UA and BP; renal consultation for patients with renal dysfunction |
| Reproductive | Impaired fertility, infertility, delayed puberty, sexual dysfunction | Tanner every 6 months; testosterone by age 14; age-appropriate sperm analysis; LH, FSH, estradiol by age 13 | 1 year and yearly thereafter | Hormone levels without specified interval | Yearly physical examination and Tanner staging; testosterone, LH, and FSH in males ≥11 years, yearly for 2 years; age- appropriate sperm analysis; LH, FSH, AMH, and estradiol in female patients ≥11 years, at 1 and 2 years post-HSCT | — |
| Organ system | Late effects | Children's Oncology Group27 | National Marrow Donor Program28 | Bhatia et al29 | SCD specific consortium30 | Fitzhugh et al31 |
|---|---|---|---|---|---|---|
| Cardiovascular | Elevated TRJV, pulmonary and systemic HTN, systolic/diastolic dysfunction, dyslipidemia, cardiac iron overload, myocardial strain | Yearly BP; lipids every 2 years; echo every 1-5 years | Regular monitoring of risk factors and complications but no specific time frame | Yearly BP; lipids every 2 years; echo every 1-5 years | Yearly BP and echo; lipids every 5 years | Yearly BP and echo; lipids every 1-2 years; cardiac MRI monitoring in those with cardiac iron overload before HSCT; cardiology/pulmonology consult for those with severe cardiac disease |
| Pulmonary | Decreased pulmonary function | PFTs at 1-year post-HSCT | PFTs at 6 mo, 12 mo, yearly | PFTs at 1-year post- HSCT | PFTs at 3, 6, and 12 mo and then yearly for 2 years; yearly PFTs for those with early compromise or chronic GVHD until off immunosuppressive medications; echo with evaluation of TRJV at 1 year; pulmonary arterial pressure if TRJV >3 m/s to confirm pulmonary HTN | PFTs at 3, 6, and 12 mo post-HSCT, then yearly; less frequent studies for patients who are not symptomatic, are free of GVHD, and have stable PFTs; pulmonary consultation for those with new or worsening abnormal PFTs |
| Renal | Hyperfiltration, proteinuria, acute/chronic renal disease, sickle cell nephropathy | Renal function for 1 year; yearly UA and BP | BP and renal function tests at 6 mo, 1 year, yearly | Renal function at 1 year; yearly UA and BP | Monitor until off nephrotoxic therapy and yearly for 2 years; UA for blood/protein at 1 year. Microalbuminuria testing yearly for 2 years; yearly BP | Monitor renal function until off nephrotoxic therapy and then yearly; less frequent testing for patients with stable electrolytes and renal function; yearly UA and BP; renal consultation for patients with renal dysfunction |
| Reproductive | Impaired fertility, infertility, delayed puberty, sexual dysfunction | Tanner every 6 months; testosterone by age 14; age-appropriate sperm analysis; LH, FSH, estradiol by age 13 | 1 year and yearly thereafter | Hormone levels without specified interval | Yearly physical examination and Tanner staging; testosterone, LH, and FSH in males ≥11 years, yearly for 2 years; age- appropriate sperm analysis; LH, FSH, AMH, and estradiol in female patients ≥11 years, at 1 and 2 years post-HSCT | — |
AMH, anti-Mullerian hormone; BP, blood pressure; echo, echocardiogram; FSH, follicle-stimulating hormone; HTN, hypertension; LH, luteinizing hormone; MRI, magnetic resonance imaging; UA, urinalysis.
Conclusion
Overall, curative therapies for SCD have transformed the course of the disease, but they are not without risks. Pursuing curative treatment should be decision that is made carefully with the guidance of highly trained providers and taking into consideration an individual patient's current risk factors and disease burden. Our understanding of long-term effects of curative therapies in patients with SCD remains limited in both adults and children and is further limited to allogeneic HSCT. It is essential that there are continued efforts to study this area to be able to educate patients and families and guide providers in their clinical care.
Acknowledgment
1U01HL156620 Clinical and genetic risk factors associated with adverse long-term health outcomes after curative therapies in individuals with sickle cell disease (COALESCE).
Conflict-of-interest disclosure
Rohini Chakravarthy: no competing financial interests to declare.
Debra L. Friedman: no competing financial interests to declare.
Off-label drug use
Rohini Chakravarthy: nothing to disclose.
Debra L. Friedman: nothing to disclose.
