

Abstract
The myelodysplastic syndromes (MDS) are a heterogeneous group of malignant hematopoietic stem cell disorders characterized by ineffective growth and differentiation of hematopoietic progenitors leading to peripheral blood cytopenias, dysplasia, and a variable risk of transformation to acute myelogenous leukemia. As most patients present with lower-risk disease, understanding the pathogenesis of ineffective hematopoiesis is important for developing therapies that will increase blood counts in patients with MDS. Various inflammatory cytokines are elevated in MDS and contribute to dysplastic differentiation. Inflammatory pathways mediated by interleukin (IL) 1b, IL-6, IL-1RAP, IL-8, and others lead to growth of aberrant MDS stem and progenitors while inhibiting healthy hematopoiesis. Spliceosome mutations can lead to missplicing of genes such as IRAK4, CASP8, and MAP3K, which lead to activation of proinflammatory nuclear factor κB–driven pathways. Therapeutically, targeting of ligands of the transforming growth factor β (TGF-β) pathway has led to approval of luspatercept in transfusion-dependent patients with MDS. Presently, various clinical trials are evaluating inhibitors of cytokines and their receptors in low-risk MDS. Taken together, an inflammatory microenvironment can support the pathogenesis of clonal hematopoiesis and low-risk MDS, and clinical trials are evaluating anti-inflammatory strategies in these diseases.
Learning Objectives
Discuss the pathologic inflammatory pathways in myelodysplastic syndromes (MDS) and their current therapeutic targeting strategies
Discuss the role of the transforming growth factor β pathway in MDS and efficacy of luspatercept in transfusion-dependent refractory anemia with ring sideroblasts
Discuss the most common pathologic spliceosome mutations in MDS that can lead to inflammation and ongoing clinical trials
CLINICAL CASE 1
A 65-year-old man with a history of hypertension and gout presented to the hematology clinic after routine bloodwork revealed anemia. A complete blood count (CBC) revealed a white blood cell (WBC) count of 4.5 × 109/µL, with an absolute neutrophil count (ANC) of 2.5 × 109/µL, hemoglobin (Hgb) of 6.2 g/µL, and platelet count of 150 K/µL. B12, folate, iron, and copper levels were normal, and his erythropoietin level was 635 mU/mL. A bone marrow (BM) biopsy was performed, showing a hypercellular marrow (60%-70% cellularity), with marked dysplastic changes in 25% of erythroid cells, 2% of blasts, and 20% of ringed sideroblasts (RSs) by iron staining; cytogenetics was normal. Next-generation sequencing (NGS) revealed SF3B1K700E mutation with 25% variant allele frequency (VAF). The patient was diagnosed with low-risk myelodysplastic syndrome (MDS) with RS and single-lineage dysplasia and started therapy with subcutaneous luspatercept 1 mg/kg every 3 weeks with transfusion support as needed. His Hgb steadily increased to 9.6 g/µL by the fifth dose of luspatercept and he became transfusion independent.
Introduction
MDS are a heterogeneous group of malignant hematopoietic stem cell disorders characterized by ineffective growth and differentiation of hematopoietic progenitors leading to peripheral blood cytopenias, dysplasia, and a variable risk of transformation to acute myelogenous leukemia. Cytopenias are the commonest presenting symptoms, and most patients have lower-risk disease. Thus, understanding the pathogenesis of ineffective hematopoiesis is important for developing therapies that will benefit most patients with MDS.
Recent experimental evidence has shown that MDS can arise from disease-initiating stem and progenitors that are enriched in mutations and cytogenetic alterations.1-5 Emerging data demonstrate that inflammation can lead to selective outgrowth of aberrant stem cells while inhibiting healthy hematopoiesis, resulting in worsening of cytopenias in MDS.6-9 As MDS is a highly heterogeneous disease, numerous proinflammatory cytokines such as tumor necrosis factor α, interferons, granulocyte colony-stimulating factor, thrombopoeitin, IL-6, transforming growth factor β (TGF-β), IL-8, IL-1, and others have been shown to be overexpressed in different human sample-based analyses.6,8,9 In fact, certain inflammatory mediators such as IL-1RAP, IL-8, IL-6, tumor necrosis factor α, and IL-1b have been shown to directly support the growth of aberrant MDS stem cells in preclinical models.6,7,10-12 We discuss some of the important pathologic inflammatory pathways in MDS and their current therapeutic targeting strategies.
Role of the TGF-β pathway in MDS
The TGF-β superfamily contains numerous growth factors that regulate hematopoietic stem cell (HSC) proliferation and differentiation. TGF-β signaling is mediated by a regulatory circuit of inhibitory and activating SMAD proteins that can inhibit the proliferation of HSCs while increasing erythroid differentiation, altogether leading to dysplastic erythropoiesis and reduced erythroid output with anemia phenotypes (Figure 1).13-15
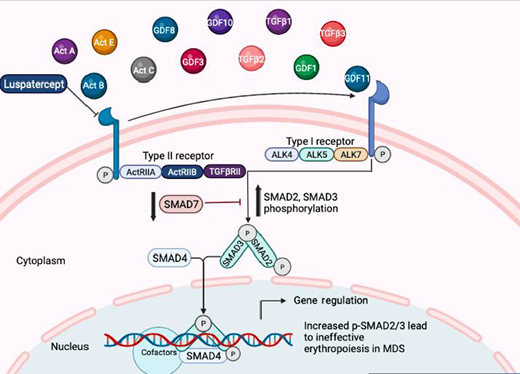
TGF-β pathways in MDS: Various ligands belonging to the TGF-β pathway bind to activin-like kinase (ALK) receptors and activate SMAD proteins. SMAD proteins act as transcription factors and regulate important hematopoietic genes that are altered in MDS. ActRIIA, activin receptor type IIA; ActRIIB, ActRIIB.
TGF-β pathways in MDS: Various ligands belonging to the TGF-β pathway bind to activin-like kinase (ALK) receptors and activate SMAD proteins. SMAD proteins act as transcription factors and regulate important hematopoietic genes that are altered in MDS. ActRIIA, activin receptor type IIA; ActRIIB, ActRIIB.
The binding of various ligands, such as TGF-β, activins, GDF11, and GDF8, lead to activation and phosphorylation of activin-like kinase-containing receptors, which, in turn, activate a regulatory circuit of SMAD proteins.14,16,17 SMADs 2/3 are phosphorylated by TGF-β and GDF8/11 ligands, and these phosphorylated SMAD proteins act as transcription factors that can regulate important cell cycle and differentiation genes, leading to altered and dysplastic hematopoiesis. SMAD2/3 activation has been seen in preclinical models of MDS as well as in primary bone marrow samples.18,19 Reduced levels of endogenous inhibitors (SMAD7 and SKI) and altered levels of microRNAs have been shown to contribute to enhanced downstream activation of the TGF-β pathway and suppression of hematopoiesis.18-21 Furthermore, patients with MDS have been shown to have higher levels of activins and GDF11 compared with healthy controls. In a mouse model of MDS, increased GDF11 levels were associated with erythroid hyperplasia and inefficient erythropoiesis.22
Ligand trap luspatercept is approved for the treatment of anemia in MDS
Luspatercept is a recombinant fusion protein derived from human activin receptor type IIb linked to a portion of immunoglobulin G. It is approved for the treatment of adults with transfusion-dependent anemia due to very low-, low-, and intermediate-risk MDS with RSs who had an unsatisfactory response to or are ineligible for erythropoietin-based therapy. Luspatercept can bind various TGF-β superfamily ligands, including GDF8, GDF11, and activin B, enhancing late-stage erythropoiesis by decreasing SMAD2/3 activation and signaling.14 In mouse models of MDS and β-thalassemia, luspatercept decreased SMAD2/3 phosphorylation, reduced erythroid hyperplasia, enhanced erythroid maturation, and increased hemoglobin levels.22,23
Luspatercept was evaluated in an open-label phase 2 trial that included 58 anemic patients with low-risk (LR)-MDS (PACE-MDS) and resulted in erythroid hematologic improvement in 38% of patients. Responses were higher in patients with spliceosome mutations (SF3B1, SRSF2, U2AF1, and ZRSR2) and with ≥15% ringed sideroblasts. Treatment was well tolerated, with only 3 grade 3 treatment-related adverse events (1 case of myalgia, 1 of increased blast cell count, and 1 case of general physical health deterioration).24 These results led to the phase 3 MEDALIST trial, which resulted in the approval of luspatercept by the US Food and Drug Administration in April 2020 and the European Medicines Agency in June 2020 for the treatment of anemia in adults with LR-MDS-RS or MDS/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis requiring ≥2 red blood cell (RBC) units/8 weeks after erythropoiesis-stimulating agent (ESA) failure. This was a double-blind, placebo-controlled trial in which 229 anemic, lower-risk, nondel(5q) patients with ≥15% RSs or ≥5% RSs with SF3B1 mutation received luspatercept or placebo. Patients were resistant to ESA (or had elevated serum erythropoietin levels at baseline). Patients treated with luspatercept had 38% RBC-transfusion-independence for >8 weeks compared with 13% in the placebo group (P < .0001), with a median duration of RBC-transfusion-independence of 30.6 weeks in the luspatercept arm. Adverse events were similar to the previously reported trial.25 There are several ongoing clinical trials evaluating luspatercept in non-RS MDS as well as in combination with other agents (Table 1).
Selected trials targeting inflammation in MDS and CH
| Class | Agent | Trial name | Phase | Population | Status | Identifier |
|---|---|---|---|---|---|---|
| TGF-β pathway | Luspatercept | PACE-MDS (24) | 2 | LR-MDS | Completed | NCT01749514 |
| TGF-β pathway | Luspatercept vs placebo | MEDALIST (25) | 3 | LR-MDS-RS | Completed | NCT02631070 |
| TGF-β pathway | Luspatercept vs epoetin alfa | COMMANDS | 3 | LR-MDS | Recruiting | NCT03682536 |
| TGF-β pathway | Luspatercept + lenalidomide | — | 1/2 | LR-MDS | Recruiting | NCT04539236 |
| TGF-β pathway | Galunisertib | — | 2 | LR-MDS | Completed | NCT02008318 |
| TGF-β pathway | Vactosertib | — | 1/2 | LR-MDS | Completed | NCT03074006 |
| TIM-3 pathway TGF-β pathway IL-1β inhibitor | Sabatolimab NIS793 Canakinumab | — | 1b | LR-MDS | Recruiting | NCT04810611 |
| IL-1β inhibitor | Canakinumab | — | 2 | LR-MDS or MDS/MPN | Recruiting | NCT05237713 |
| IL-8 inhibitor | BMS-986253 ± PO decitabine and cedazuridine | — | 1/2 | HR-MDS with prior HMA therapy or LR-MDS with cytopenias | Not yet Recruiting | NCT05148234 |
| CXCR1 and CXCR2 inhibitor | SX-682 | — | 1 | MDS with disease progression or prior therapy intolerance | Recruiting | NCT04245397 |
| TLR2 inhibitor | Tomaralimab | — | 1/2 | LR-MDS with prior HMA | Completed | NCT02363491 |
| IRAK4 inhibitor | Emavusertib (CA-4948) | CURIS | 1 | HR-MDS and AML | Active | NCT04278768 |
| IRAK4 inhibitor | Emavusertib (CA-4948) | LUCAS | 2 | LR-MDS | Recruiting | NCT05178342 |
| Class | Agent | Trial name | Phase | Population | Status | Identifier |
|---|---|---|---|---|---|---|
| TGF-β pathway | Luspatercept | PACE-MDS (24) | 2 | LR-MDS | Completed | NCT01749514 |
| TGF-β pathway | Luspatercept vs placebo | MEDALIST (25) | 3 | LR-MDS-RS | Completed | NCT02631070 |
| TGF-β pathway | Luspatercept vs epoetin alfa | COMMANDS | 3 | LR-MDS | Recruiting | NCT03682536 |
| TGF-β pathway | Luspatercept + lenalidomide | — | 1/2 | LR-MDS | Recruiting | NCT04539236 |
| TGF-β pathway | Galunisertib | — | 2 | LR-MDS | Completed | NCT02008318 |
| TGF-β pathway | Vactosertib | — | 1/2 | LR-MDS | Completed | NCT03074006 |
| TIM-3 pathway TGF-β pathway IL-1β inhibitor | Sabatolimab NIS793 Canakinumab | — | 1b | LR-MDS | Recruiting | NCT04810611 |
| IL-1β inhibitor | Canakinumab | — | 2 | LR-MDS or MDS/MPN | Recruiting | NCT05237713 |
| IL-8 inhibitor | BMS-986253 ± PO decitabine and cedazuridine | — | 1/2 | HR-MDS with prior HMA therapy or LR-MDS with cytopenias | Not yet Recruiting | NCT05148234 |
| CXCR1 and CXCR2 inhibitor | SX-682 | — | 1 | MDS with disease progression or prior therapy intolerance | Recruiting | NCT04245397 |
| TLR2 inhibitor | Tomaralimab | — | 1/2 | LR-MDS with prior HMA | Completed | NCT02363491 |
| IRAK4 inhibitor | Emavusertib (CA-4948) | CURIS | 1 | HR-MDS and AML | Active | NCT04278768 |
| IRAK4 inhibitor | Emavusertib (CA-4948) | LUCAS | 2 | LR-MDS | Recruiting | NCT05178342 |
AML, acute myelogenous leukemia; HMA, hypomethylating agent; MPN, myeloproliferative neoplasms; TBD, to be determined; TLR2, Toll-like receptor 2.
CLINICAL CASE 2
A 73-year-old woman with a history of hypertension, diabetes, and previously diagnosed low-risk MDS was referred to our clinic for a second opinion after 7 months of treatment with ESA for symptomatic anemia, with an initial response leading to transfusion independence, but now with loss of response. A CBC revealed a WBC count of 3.5 × 109/µL, with an ANC of 2.0 × 109/µL, Hgb of 6.0 g/µL, and a platelet count of 90 K/µL. A BM biopsy was performed, showing a hypercellular marrow (70%-80% cellularity), with marked dysplastic changes in 30% of erythroid cells and 2% of blasts; cytogenetics was normal. NGS identified a U2AF1 S34F mutation with 19% VAF, confirming the diagnosis of LR-MDS.
Spliceosome mutations in MDS can lead to inflammation
Mutations in RNA splicing genes are commonly seen in MDS, accounting for 40% to 65% of cases. Commonly mutated genes include SF3B1 (20%-25%), SRSF2 (12%), U2AF1 (10%), and ZRSR2 (2%-6%).26
The posttranslational process of removing introns and ligating exons to generate mature transcripts appropriate for translation is known as pre–messenger RNA splicing. Before a transcript can be exported to the cytoplasm for translation, introns must be removed, a process carried out by the spliceosome. Dysregulated pre–messenger RNA splicing may result in functional alterations in the translated protein, possibly resulting in the creation of oncogenes or the inactivation of tumor suppressor genes. Various studies have shown that misspliced versions of numerous important genes can be seen in primary patient samples and in spliceosome mutant MDS models. A few of these misspliced genes include MAP3K7, CASP8, GNAS, and IRAK4, all of which can activate the proinflammatory nuclear factor κB (NF-κB) and other proliferative pathways (Figure 2).27-30 Recent data show that loss of function of the helicase DDX41, a gene that is germline mutated in MDS, can also lead to increased RNA: DNA hybrids and activates the inflammatory cGAS-STING pathway.31

Spliceosome mutations and inflammation in MDS: Mutations in splicing genes can lead to expression of isoforms that lead to maximal activation of proinflammatory NF-κB–driven pathways in MDS.
Spliceosome mutations and inflammation in MDS: Mutations in splicing genes can lead to expression of isoforms that lead to maximal activation of proinflammatory NF-κB–driven pathways in MDS.
Interleukin 1 receptor–associated kinase 4 (IRAK4) is an important component of the innate immune signaling cascade that is activated by the MyD88 adapter protein downstream of Toll-like receptors. This in turn results in stimulation of NF-κB, prompting inflammatory responses, oncogenesis, and survival of malignant cells.12 IRAK4 can exist in longer and shorter isoforms, and U2AF1 and SF3B1 mutations can lead to altered exon inclusion, leading to preferential production of a longer isoform (IRAK4-L). This longer isoform contains an N-terminal death domain and C-terminal kinase domain that assemble stochastically with the Myddosome complex, resulting in maximal activation of innate immune signaling pathways.30 Preclinical studies have shown that inhibition of IRAK4-L by pharmacologic and genetic means can suppress leukemic proliferation both in vitro and in vivo. Thus, U2AF1 and SF3B1 mutations induce the expression of therapeutically targetable active IRAK4 isoforms and establish a link between a mutation and chronic innate immune signaling activation in MDS.30 Clinical trials are now evaluating the efficacy of IRAK4 inhibitors in MDS (Table 1).
CLINICAL CASE 3
A 55-year-old man with a history of iron-deficiency anemia was initially referred to our clinic in 2007 for iron-deficiency anemia secondary to a duodenal ulcer that had been diagnosed when he presented to the hospital with Hgb of 5.8 g/µL, mean corpuscular volume of 68 fL, ferritin of 4 ng/mL, iron of 15 µg/dL, saturation of 5%, and total iron-binding capacity of 515 µg/dL. He was started on proton pump inhibitors, and his Hgb recovered to 13 g/µL; at the time, a peripheral blood NGS panel had been obtained, which had revealed a TET2 mutation (VAF of 12%). He is a nonsmoker who resides in New York City with his wife and worked as a firefighter during the World Trade Center (WTC) terrorist attacks. He was referred again to our clinic for worsening exertional dyspnea; his current CBC shows a WBC count of 3.7 × 109/µL, with an ANC of 2.2 × 109/µL, Hgb of 6.4 g/µL, and a platelet count of 100 K/µL. A BM biopsy was performed, showing a hypercellular marrow (50%-60% cellularity), with dysplastic changes in 15% of erythroid cells and 2% blasts; cytogenetics was normal. NGS identified a TET2 mutation with 22% VAF, and the evolution from clonal hematopoiesis (CH) to low-risk MDS was confirmed.
Inflammation is associated with increased clonal hematopoiesis
CH is defined as the acquisition of somatic mutations in blood cells in individuals without a hematologic malignancy, dysplasia, or cytopenia. CH increases the risk of leukemia by 0.5% to 1.0% per year, and it is associated with higher morbidity and mortality from a variety of inflammatory conditions and atherosclerotic cardiovascular disease.32-34 Furthermore, the development of CH is associated with smoking, increasing age, and exposure to genotoxic stimuli.35 Aging is associated with the development of CH potentially caused by an increased inflammatory milieu seen in the bone marrow.6-9,36 Several proinflammatory cytokines are elevated in preclinical animal models of aging that are detrimental to normal HSCs and promote the growth of aberrant clones.
As illustrated in case 3, several exposures are associated with a higher risk of developing CH. First responders to the WTC 9/11 disaster were exposed to proinflammatory particulate matter containing high levels of potential carcinogens, and a recent study identified a significantly higher proportion of WTC- exposed first responders with CH (10%) vs non-WTC-exposed firefighters (6.7%; P = .0006). The frequency of somatic mutations in WTC-exposed first responders showed an age-related increase and predominantly affected DNMT3A, TET2, and other CH-associated genes. Other studies have also shown that systemic inflammation can increase the size of the mutant clone in preclinical CH models and also potentially accelerate the transformation to overt cytopenias.6-9,36
CH mutations, especially in the TET2 and JAK2 genes, can by themselves lead to a proinflammatory state by making macrophages more proliferative and secretory.37,38 Other myeloid-derived inflammatory mediators have also been shown to be elevated in MDS. S100A8/S100A9 are cytosolic alarmins predominantly secreted by myeloid cells, which activate the inflammasome via the Toll-like receptor 4 and NLRP3 pathways and are found to be elevated in MDS.39 The binding of the S100A8/S100A9 heterodimer to the CD33 receptors on myeloid-derived suppressor cells also leads to their expansion, leading to MDS-like phenotypes in preclinical models.39 S100A8/9 have also been shown to be associated with anemia seen in MDS with 5q deletions in preclinical models.40 Based on these studies, inhibition of inflammasome signaling is emerging as a potential target in MDS and CH and is being investigated in trials (Table 1).
MDS and autoimmunity
MDS and chronic myelomonocytic leukemia have been linked to autoimmune and inflammatory diseases such as polychondritis, vasculitis, and arthritis. Retrospective studies have postulated that more than 10% of patients with MDS may have concomitant autoimmune/inflammatory diseases with enrichment of TET2, IDH, and SRSF2 mutations.41 In fact, azacitidine has demonstrated efficacy in the treatment of autoimmune disorders associated with MDS/chronic myelomonocytic leukemia in small studies and suggests that further studies are warranted to test these associations.42 Last, an inflammatory syndrome that can overlap with MDS, the VEXAS (vacuoles, E1 enzyme, x-linked, autoinflammatory, somatic) syndrome, has been recently described; it is characterized by an often fatal, treatment-refractory inflammatory disorder that develops in late adulthood. The syndrome is characterized by fevers, cytopenias, characteristic vacuoles in myeloid and erythroid precursor cells, dysplastic bone marrow, neutrophilic cutaneous and pulmonary inflammation, chondritis, and vasculitis. Most of these patients meet clinical criteria for an inflammatory syndrome or a hematologic disease (MDS or multiple myeloma) or both. These patients have mutations in more than 50% of their hematopoietic stem cells, including peripheral blood myeloid cells, and harbor mutations affecting p.Met41, resulting in the loss of the canonical cytoplasmic isoform of UBA1 and in the expression of a novel, catalytically impaired isoform initiated at p.Met67. These mutant peripheral blood cells show decreased ubiquitylation and activated innate immune pathways.43
Taken together, these emerging data demonstrate the role of inflammation in perpetuating MDS pathobiology. It is hoped that strategies targeting specific inflammatory cascades will lead to improvements in cytopenias and decrease the risk of leukemia progression in MDS.
Conflicts-of-interest disclosure
Amit Verma has received research funding from Prelude, BMS, GSK, Incyte, Medpacto, Curis, and Eli Lilly; is a scientific advisor for Stelexis, Bakx, Curis, Novartis, Acceleron and Celgene; receives honoraria from Stelexis and Janssen; and holds equity in Stelexis, Bakx, and Throws Exception.
Jesus D. Gonzalez-Lugo has no competing financial interests to declare.
Off-label drug use
Jesus D. Gonzalez-Lugo: use of investigational agents in clinical trials is discussed.
Amit Verma: use of investigational agents in clinical trials is discussed.
*Note
The article “Targeting inflammation in lower-risk MDS” beginning on page 382 is not eligible for continuing medical education (CME) credit.
