Background
Pakistan is among the countries with a high prevalence of β-thalassemia, with a β-thalassemia gene frequency of 5% to 8% and ∼5000 new cases diagnosed each year. In Pakistan, the mean annual expenditure for managing disease in a child with thalassemia is US$4500, which is much more than the per capita income in Pakistan.
Objectives
To prevent thalassemia major (TM) by extended family screening and chorionic villus sampling (CVS).
To produce a national protocol for iron chelation and safe blood transfusion in patients with TM.
To investigate hemoglobin F (HbF) augmenting agent in TM patients to reduce the requirement for a blood transfusion.
To discover primary and secondary genetic modifiers as a primary screening test for treatment with HbF augmenting agent.
To establish the role of proteomics and metabolites as markers for response to hydroxyurea (HU) therapy.
To establish bone marrow transplantation (BMT) centers throughout Pakistan to cure TM.
Material and Methods
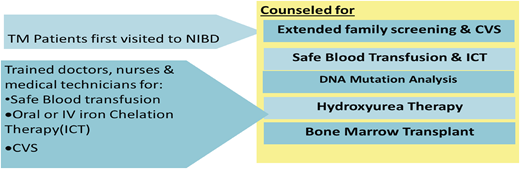
Capacity Building
Doctors, nurses, laboratory technicians, and blood banking staff were trained.
A dedicated research department was built whose staff consisted of clinical research associates, study coordinators, and an epidemiologist.
Hypothesis
What began with a strong vision in 2001, became a project to establish safe blood banking, iron chelation therapy (ICT), and curative treatment (ie, BMT) throughout Pakistan.
Evidence-based management will lead to reduction and eventually eradication of thalassemia in Pakistan.
Thalassemia prevention program
Effectiveness and feasibility of CVS for prenatal diagnosis of β-thalassemia in a Muslim-majority community in Pakistan
In a total of 798 fetuses, 224 (28%) were diagnosed as having TM, 400 (50.1%) as having thalassemia minor, 173 (21.6%) as being healthy, and 1 (0.12%) as having an undetected mutation. Procedure-related complications were seen in 20 fetuses (2.4%), and missed abortion occurred in 6 of the 798 fetuses. Seven couples (3%) refused to abort a fetus with ΤΜ, whereas 97% of fetuses were aborted according to recommendations.1
Molecular epidemiology of β-thalassemia in Pakistan.
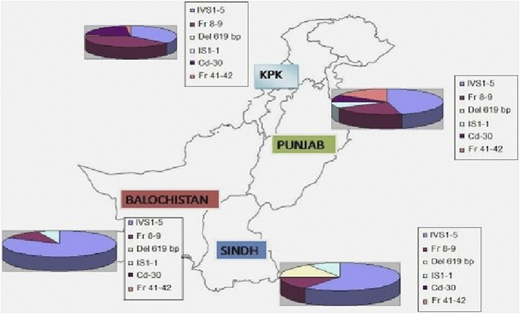
Over a 5-year period, DNA from 648 blood samples (including specimens from CVS) were analyzed for the 12 most common β-thalassemia mutations found in the Pakistani population by using a multiplex amplification refractory mutation system.2 Distribution of different genetic mutations in different provinces is shown in Figure 1.
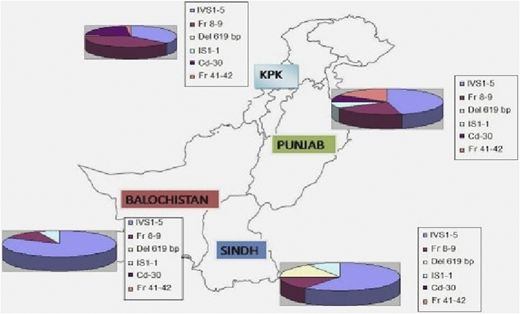
Distribution of different genetic mutations in different provinces. KPK, Karachi, Pakistan.
Distribution of different genetic mutations in different provinces. KPK, Karachi, Pakistan.
Efficacy of HU in reducing the packed red cell (PRC) transfusion requirement among children having BMT (Karachi HU Trial [KHUT]).
HU was offered to 23 patients who showed significant reduction in the volume of packed red blood cells transfused (from 2126.45 to 1489.59 mL).3
Efficacy of HU in providing transfusion independence in β-thalassemia.
One hundred fifty-two patients (41%) receiving HU therapy showed complete response (CR or transfusion independence), whereas partial response (PR, defined as a 50% reduction in the requirement for blood transfusion) was observed in 39% of patients.4
Metabolite profiling of thalassemic patients.
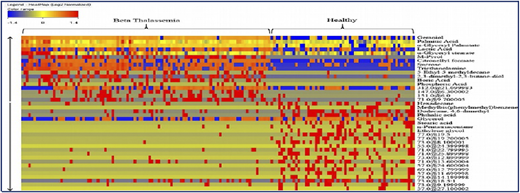
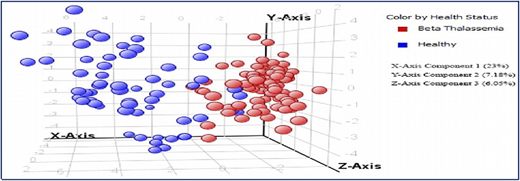
A collaborative study with the International Center for Chemical and Biological Sciences (ICCBS) was performed using metabolite profiling of 161 serum samples from healthy volunteers (n = 61) and from patients with β-thalassemia (n = 100); the profiling used gas chromatography-electron ionization-mass spectrometry.5 After using gas chromatography-mass spectrometry to analyze the samples, metabolites were identified by using Agilent Mass Hunter Qualitative Analysis software and the National Institute of Standards and Technology library. Forty metabolites (Figure 2) were identified as being significantly different between the 2 groups at a probability of 0.05 and a fold change >1.5. Figure 3 shows a plot of principle components analysis (PCA) scores of healthy participants and patients with TM.
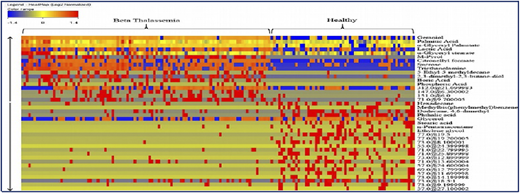
Forty significant metabolites that showed significant differences among healthy individuals and patients with β-thalassemia.
Forty significant metabolites that showed significant differences among healthy individuals and patients with β-thalassemia.
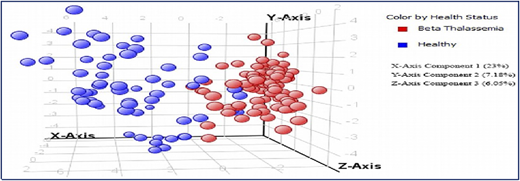
Plot of principle components analysis (PCA) scores of healthy individuals and patients with TM.
Plot of principle components analysis (PCA) scores of healthy individuals and patients with TM.
Pharmacoproteomics profiling of β-thalassemia patients in response to treatment with HU.
In this study, we performed comparative analysis of plasma proteomes in transfusion-dependent children (n = 10) who had ΤΜ before and after treatment with HU as well as responders vs nonresponders to HU treatment.6 Plasma was collected before and after 6 months of HU treatment, and patients were subcategorized on the basis of their response to HU. Among 400 samples, 28 proteins were found to be significantly different in patients stratified into 2 groups: before being treated with HU and after being treated with HU.
Comparison of conditioning regimens in β-thalassemia patients undergoing hematopoietic stem cell transplantation.
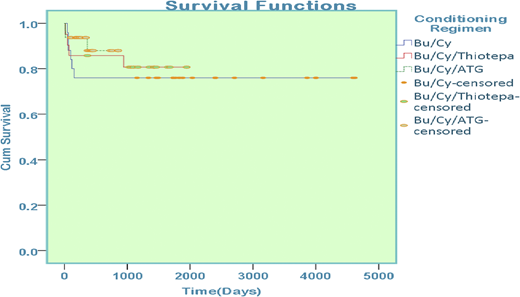
Patients with thalassemia who underwent hematopoietic stem cell transplantation were enrolled in the study and were stratified according to the conditioning regimen offered. Of 78 patients, 25 received busulfan-cyclophosphamide, 21 received busulfan-cyclophosphamide-thiotepa, and 43 patients were offered treatment with busulfan-cyclophosphamide-antithymocyte globulin.
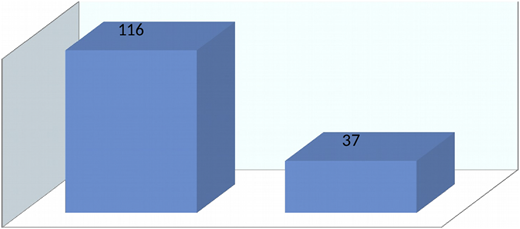
Sex mismatch was present in 45 patients (50.5%) who underwent transplantation. Major ABO mismatch was observed in 9 patients (11.5%) and minor ABO mismatch was observed in 7 patients (9%). Comparison of survival among groups reported 76% in those treated with busulfan-cyclophosphamide, 81% in those treated with busulfan-cyclophosphamide-thiotepa, and 86% in those treated with combination busulfan-cyclophosphamide-antithymocyte globulin; the difference was statistically significant (P = .008) (Figure 4).
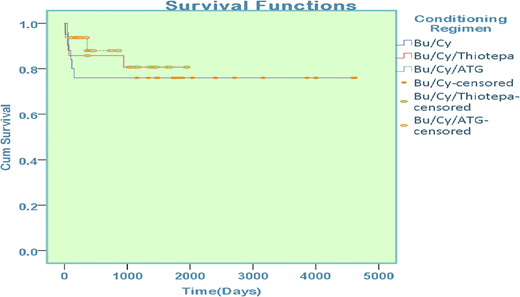
Overall survival among patients with thalassemia on the basis of conditioning regimen.
Overall survival among patients with thalassemia on the basis of conditioning regimen.
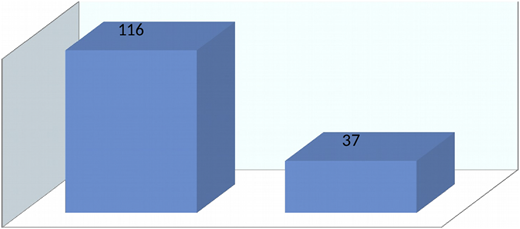
Overall, transplants in thalassemia patients (n = 153) showed 76% survival at the National Institute of Blood Diseases and Bone Marrow Transplantation (NIBD) (Figure 5).
Results
Thalassemia prevention program
As a part of the thalassemia prevention program, 2458 CVS procedures were performed. In all, 624 fetuses (25%) were diagnosed as having TM, 1253 (51%) had the sickle cell trait, and 2 (0.08%) had an undetected mutation. Five hundred families and >5000 individuals were screened to determine whether they were thalassemia carriers. Pakistan has instituted a law that mandates screening for thalassemia before marriage.
Better supportive therapy
Uniform guidelines have been made for blood screening and ICT through the Thalassemia Federation of Pakistan. An ongoing study showed reduction in the level of serum ferritin with deferiprone monotherapy, with a combination of deferasirox and deferoxamine, and with a combination of deferiprone and deferoxamine compared with deferasirox monotherapy.
HbF augmenting therapy
Between 2003 and October 2014, 1135 patients were enrolled on the study, 165 were dropped because of noncompliance, and 56 did not receive HU for >6 months.
Of the remaining 914 patients, 315 (34.4%) achieved CR, 365 (40.0%) achieved a PR, and 234 (25.6%) had no response.
HU is now used by pediatricians and hematologists throughout Pakistan, and >10 000 patients have benefitted: half of those have shown a reduction in the requirement for a blood transfusion, and half the children did not require any further blood transfusions.
BMT activity
From 2001 to 2018, 5 BMT units have been established throughout the country that have the capacity to perform 30 to 40 BMTs for thalassemia per month.
Conclusion
The initiative that began in 2001 has shown slow but steady progress. Uniform progress has been made in training on ways to prevent thalassemia, providing better supportive care with safe blood and ICT, managing thalassemia without blood transfusion with HbF augmenting agent, and offering curative therapy by establishing BMT units throughout the country. Our progress is encouraging, but additional national and international collaborations are needed to find cost-effective ways to manage and prevent thalassemia in developing countries.
Acknowledgements
The authors thank the BMT staff, the TM patients for consenting to be included in this study, ICCBS, the National Center for Proteomics, and the University of Karachi.
Authorship
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Saqib Hussain Ansari, Consultant Hematologist, National Institute of Blood Disease and Bone Marrow Transplantation, ST 2/A, Block 17, Gulshan-e-Iqbal, KDA scheme 24, Karachi, Pakistan.