

Key Points
One third of patients with USS have a neonatal episode of severe hemolytic jaundice with thrombocytopenia induced by an unknown trigger.
A USS patient with persisting PDA and recurrent neonatal hemolysis/thrombocytopenia suggests blood flow through the PDA as trigger.
Introduction
Upshaw-Schulman syndrome (USS) or congenital thrombotic thrombocytopenic purpura (TTP) is a rare but potentially fatal thrombotic microangiopathy caused by a severe deficiency of ADAMTS13 activity resulting from biallelic ADAMTS13 gene mutations.1-5 Approximately 25% to 40% of patients with USS have a neonatal episode of severe hemolytic jaundice with thrombocytopenia that requires exchange blood transfusion, which is the classic hallmark of USS.6-11 This hallmark is distinct from the neonatal alloimmune Coombs-positive hemolytic jaundice caused by feto-maternal ABO or more often Rhesus (Rh) incompatibility that begins during pregnancy. Some patients with USS have onset of disease only during later childhood, adolescence, or even adulthood, suggesting that severe deficiency of ADAMTS13 activity is compatible with life as long as no additional triggers such as infections occur. Thus far, it is unclear what triggers neonatal hemolysis and thrombocytopenia in a substantial proportion of USS patients. The following observation of an exceptional case strongly suggests that severe neonatal hemolytic jaundice and thrombocytopenia in USS is triggered by the blood flow conditions in the ductus arteriosus that is normally patent for the first 48 hours after birth.
Methods
USS patients
In the registry of Nara Medical University (as of June 2019), 22 (36.6%) of 60 patients with USS whose biallelic ADAMTS13 gene mutations had been identified also had detailed histories on their neonatal period available and had an episode of the classic hallmark necessitating exchange blood transfusion within the first week after birth (Table 1). The exceptional course of 1 newborn with USS (No. 16 in Table 1) led to the hypothesis outlined in this article. This study was conducted with the approval of the ethical committee of Nara Medical University. Written informed consent was obtained from the patient’s parents.
ADAMTS13 analysis
Results and discussion
As shown in Table 1, 21 of the 22 patients with USS who needed exchange blood transfusion during the neonatal period because of severe Coombs-negative hemolytic jaundice and thrombocytopenia had a very severe deficiency of ADAMTS13 activity (<0.5% of normal); the remaining patient (No. 22) had a strongly decreased but measurable residual ADAMTS13 activity of 3.1%. Four patients showed homozygous and 18 showed compound heterozygous ADAMTS13 gene mutations that were spread throughout the entire molecule (Table 1), which was distinct compared with recent findings by Alwan et al10 who reported mutations predominantly in the pre-spacer domains in patients with childhood onset USS.
As is also evident from the synopsis of the 22 newborns with USS who needed exchange blood transfusions, most had only 1 hemolytic attack that occurred soon after birth, with the notable exception of patient No.16 who experienced 3 distinct bouts of hemolysis and thrombocytopenia during the first 30 days of life (Table 1; Figure 1A). A firm diagnosis of USS in this girl was made when she was 5 years old by the following test results: ADAMTS13 activity <0.5% of normal, ADAMTS13 antigen <0.1%, no circulating inhibitor of ADAMTS13, and the compound heterozygous ADAMTS13 gene mutations p.Q449*/p.Q1374Sfs*22.14 The distinctive neonatal course with recurring hemolytic attacks (Figure 1A) was initially overlooked.14 She was born at full-term by vaginal delivery with the assistance of a vacuum extractor, and she weighed 3018 g. Nineteen hours after birth, she developed severe hemolytic jaundice and thrombocytopenia (first episode shown as (1) in Figure 1A; laboratory data are provided in Table 1). She underwent 4 exchange blood transfusions within the first 2 days after birth and recovered. Then, unexpectedly her general health gradually deteriorated, she developed generalized edema, and on day 8 a physician noticed a systolic cardiac murmur; cardiomegaly (cardiothoracic ratio of 0.62 on radiograph film) was documented. An echocardiogram revealed the presence of a patent ductus arteriosus (PDA) with a diameter of 3.8 mm. Subsequently, on day 11, the patient became cyanotic as a result of left cardiac failure caused by high output, and she showed recurring hemolysis and thrombocytopenia (second episode shown as (2) in Figure 1A). She was intubated and ventilated to improve oxygenation. After clinical improvement, 3 intravenous doses of indomethacin, a cyclooxygenase inhibitor that reduces plasma levels of the vasodilatory prostacyclin (PGI2), were applied with the intent of occluding the PDA. This treatment was not effective, and her clinical condition worsened with a new bout of hemolysis and decreased platelet count (third episode on day 26, shown as (3) in Figure 1A). Therefore, she underwent surgical ligation of the PDA on day 29. Thereafter, hemolysis and thrombocytopenia ceased completely until she was 14 months old at which point she had another bout of TTP along with chickenpox.
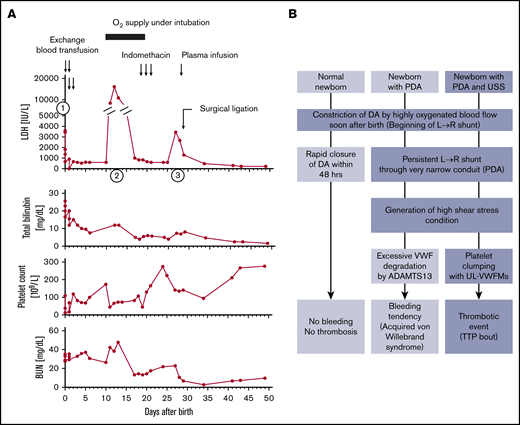
Clinical course of a patient with USS (no. 16) and schematic illustration how a USS newborn with a PDA develops hemolytic jaundice and thrombocytopenia during the newborn period. (A) First episode (indicated by ①: She was born at full-term and weighed 3018 g. Nineteen hours after birth, she developed severe hemolytic jaundice and thrombocytopenia (patient no.16 in Table 1). She underwent exchange blood transfusions 4 times within the first 2 days of life. Then, she gradually developed generalized edema, and on day 8, the comorbidity of a PDA (3.8 mm in diameter) was diagnosed. Second episode (indicated by ②: On day 11, the patient developed cyanosis as a result of left cardiac failure followed by hemolysis and thrombocytopenia, so she received oxygen therapy under intubation. Third episode (indicated by ③: After clinical improvement, on days 19 to 21 she underwent intravenous administration of indomethacin with the intention of supporting PDA closure. This treatment was ineffective, and her clinical condition worsened with recurring hemolysis and thrombocytopenia on day 26. Surgical ligation of the PDA: On day 29, she underwent surgical ligation of the PDA. Thereafter, the clinical signs of hemolysis and thrombocytopenia ceased until she was 14 months old, when she had chickenpox. (B) Schematic illustration shows how a newborn with USS who has a PDA develops severe and recurring episodes of hemolytic jaundice and thrombocytopenia during the newborn period. Note that a newborn with normal ADAMTS13 activity and persisting PDA often develops acquired von Willebrand syndrome.19,20 BUN, blood urea nitrogen; LDH, lactate dehydrogenase; UL-VWFMs, unusually large VWF multimers.
Clinical course of a patient with USS (no. 16) and schematic illustration how a USS newborn with a PDA develops hemolytic jaundice and thrombocytopenia during the newborn period. (A) First episode (indicated by ①: She was born at full-term and weighed 3018 g. Nineteen hours after birth, she developed severe hemolytic jaundice and thrombocytopenia (patient no.16 in Table 1). She underwent exchange blood transfusions 4 times within the first 2 days of life. Then, she gradually developed generalized edema, and on day 8, the comorbidity of a PDA (3.8 mm in diameter) was diagnosed. Second episode (indicated by ②: On day 11, the patient developed cyanosis as a result of left cardiac failure followed by hemolysis and thrombocytopenia, so she received oxygen therapy under intubation. Third episode (indicated by ③: After clinical improvement, on days 19 to 21 she underwent intravenous administration of indomethacin with the intention of supporting PDA closure. This treatment was ineffective, and her clinical condition worsened with recurring hemolysis and thrombocytopenia on day 26. Surgical ligation of the PDA: On day 29, she underwent surgical ligation of the PDA. Thereafter, the clinical signs of hemolysis and thrombocytopenia ceased until she was 14 months old, when she had chickenpox. (B) Schematic illustration shows how a newborn with USS who has a PDA develops severe and recurring episodes of hemolytic jaundice and thrombocytopenia during the newborn period. Note that a newborn with normal ADAMTS13 activity and persisting PDA often develops acquired von Willebrand syndrome.19,20 BUN, blood urea nitrogen; LDH, lactate dehydrogenase; UL-VWFMs, unusually large VWF multimers.
In normal newborns, the closure of the ductus arteriosus (DA) is accomplished via a multistep process15-18 : First, vasoconstriction by smooth muscle cell contraction is initiated immediately after birth when reversal of the fetal blood flow direction results in highly oxygenated blood flowing from the aorta through the DA to the pulmonary artery (left→right shunt). This first step of vasoconstriction, facilitated by a postnatal decline of the vasodilatory prostaglandin E2, leads to local endothelial injury and detachment and von Willebrand factor–mediated platelet adhesion, activation, and aggregation. Finally, anatomic remodeling results in permanent obliteration of the fetal conduit. Studies in mice16 and preterm human newborns,16,17 the latter being at high risk of persisting PDA, showed the importance of a sufficient platelet count for prompt DA closure. A PDA on day 7 was seen in 47% of preterm neonates with thrombocytopenia of <100 × 109/L,17 whereas in full-term newborns with normal platelet counts, DA closure is usually achieved within 48 hours after birth.
A persisting PDA in a normal newborn may result in acquired von Willebrand syndrome caused by excessive ADAMTS13-induced proteolysis of von Willebrand factor (VWF) subunits under the high shear stress conditions in the PDA, which results in a loss of high-molecular-weight VWF multimers.19,20 This mechanism of VWF degradation leading to acquired von Willebrand syndrome is similar to the more common situation in severe aortic stenosis termed Heyde syndrome.21 A newborn with USS who lacks any ADAMTS13 activity will typically have a first bout of unusually large VWF-induced platelet clumping with resulting thrombocytopenia and microangiopathic hemolysis with jaundice, probably triggered by the initially open DA (Figure 1B). After physiologic closure within 48 hours of life, spontaneous or exchange blood transfusion–induced remission from hemolysis and thrombocytopenia may ensue, as is commonly reported in USS patients10,11 and as was the case for 21 of the 22 carefully documented USS newborns from our Registry (Table 1). The observation of recurring bouts of thrombocytopenia and hemolytic jaundice within the first month after birth in our full-term USS newborn (No. 16 in Table 1; Figure 1A) with a persisting PDA and complete remission after surgical ligation of the high shear stress-inducing vascular conduit is strong evidence that the typical neonatal hemolytic jaundice and thrombocytopenia observed in a significant proportion of USS patients is triggered by the blood flow conditions in the DA that is open for the first 48 hours after birth.
Acknowledgments
This work was supported by research grants from the Ministry of Health, Labour, and Welfare of Japan and by the Takeda Medical Foundation.
Authorship
Contribution: Y.F. designed the study, analyzed the data, and wrote the paper; B.L., K.S., Y.T., and M.M. analyzed the data and wrote the paper; and S. Tanabe, T.K., K.K., T.M., and S. Taniguchi performed the experiments.
Conflict-of-interest disclosure: Y.F. and M.M. are inventors of an enzyme-linked immunosorbent assay (ELISA) for assessing ADAMTS13 activity. The remaining authors declare no competing financial interests.
Correspondence: Masanori Matsumoto, Nara Medical University, Shijo-cho 840, Kashihara, Nara 634-8522, Japan; e-mail: mmatsumo@naramed-u.ac.jp.


