Key Points
HA-specific iPSC-derived ECs overexpress full-length F8 after genetic engineering via a piggyBac transposon system.
Bioengineered microvascular grafts deliver full-length FVIII into the bloodstream and restore hemostasis in hemophilic SCID mice.

Abstract
Hemophilia A (HA) is a bleeding disorder caused by mutations in the F8 gene encoding coagulation factor VIII (FVIII). Current treatments are based on regular infusions of FVIII concentrates throughout a patient’s life. Alternatively, viral gene therapies that directly deliver F8 in vivo have shown preliminary successes. However, hurdles remain, including lack of infection specificity and the inability to deliver the full-length version of F8 due to restricted viral cargo sizes. Here, we developed an alternative nonviral ex vivo gene-therapy approach that enables the overexpression of full-length F8 in patients’ endothelial cells (ECs). We first generated HA patient–specific induced pluripotent stem cells (HA-iPSCs) from urine epithelial cells and genetically modified them using a piggyBac DNA transposon system to insert multiple copies of full-length F8. We subsequently differentiated the modified HA-iPSCs into competent ECs with high efficiency, and demonstrated that the cells (termed HA-FLF8-iECs) were capable of producing high levels of FVIII. Importantly, following subcutaneous implantation into immunodeficient hemophilic (SCID-f8ko) mice, we demonstrated that HA-FLF8-iECs were able to self-assemble into vascular networks, and that the newly formed microvessels had the capacity to deliver functional FVIII directly into the bloodstream of the mice, effectively correcting the clotting deficiency. Moreover, our implant maintains cellular confinement, which reduces potential safety concerns and allows effective monitoring and reversibility. We envision that this proof-of-concept study could become the basis for a novel autologous ex vivo gene-therapy approach to treat HA.
Introduction
Hemophilia A (HA) is an inherited X-chromosome–linked bleeding disorder caused by mutations in the F8 gene encoding coagulation factor VIII (FVIII).1 Approximately 1 in 5000 men are born with HA, and patients with the severe disease phenotype (∼60%) present frequent spontaneous bleeds into joints and soft tissues, which can lead to serious complications and even death.2 Current treatments for HA patients are infusions of FVIII concentrates.3 However, patients require repeated IV injections of the factor multiple times per week throughout life, which creates continuous discomfort, augments morbidity, and impairs overall quality of life.4,5 Moreover, prophylaxis for severe patients involves injections of FVIII concentrates every other day, and adherence is a constant challenge.6,7 Therefore, there is a clinical need for new approaches to treating HA, and gene therapy remains a particularly appealing alternative.8,9
Most preclinical studies of HA gene therapy have focused on the use of viral vectors, including adenovirus10,11 and adeno-associated virus (AAV).12-14 However, F8 is a relatively large gene (∼7.0 kb complementary DNA [cDNA]), and thus it cannot be effectively packaged into most existing viral vectors.15 Consequently, most efforts in HA gene therapy have been conducted with a truncated version of FVIII that lacks the B domain (referred to as BDD-FVIII). Nevertheless, mounting evidence indicates that although the B domain is not essential for coagulation, it is involved in multiple critical posttranslational functions, including FVIII secretion into the bloodstream and its later clearance from plasma.16-18 Thus, the interest in alternative gene-therapy strategies compatible with introducing the full-length version of FVIII remains.
Most gene-therapy efforts in HA have focused on direct in vivo gene delivery, mainly through AAV-mediated and liver-directed approaches. Alternatively, some studies have resorted to ex vivo gene-therapy strategies, that is, gene editing the target cells ex vivo and then transplanting these modified cells back into the patients in an effort to avoid direct use of viruses in vivo. However, notwithstanding recent advancements achieved with hemopoietic stem cells (HSCs),19-21 ex vivo gene-therapy approaches for HA have generally been challenging, in part due to the difficulty of achieving stable engraftment, with translational concerns over scalability and cell source.22,23
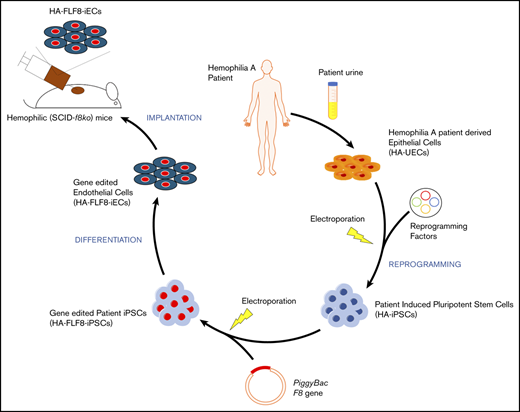
Here, we developed a novel nonviral ex vivo gene-therapy approach to overexpress full-length FVIII in endothelial cells (ECs) derived from HA patients. To this end, we generated induced pluripotent stem cells (iPSCs) from severe HA patients (HA patient–specific induced pluripotent stem cells [HA-iPSCs]) and used a piggyBac DNA transposon system to insert the full-length version of the human F8 gene into the genome of these patients’ cells. We then differentiated the full-length F8 gene-edited HA-iPSCs into competent FVIII-expressing ECs (termed HA-FLF8-iECs) with high efficiency, and demonstrated that these modified HA-FLF8-iECs could form FVIII-secreting vascular networks within subcutaneous implants in hemophilic mice, restoring therapeutic levels of FVIII activity.
Methods
Generation of patient HA-iPSCs and HA-iECs
Deidentified urine samples were obtained from patients with severe HA and from healthy individuals in accordance with institutional review board–approved protocols at Boston Children’s Hospital. Informed consent was obtained from all donors. Urine-derived epithelial cells were isolated from 7 patients (supplemental Table 1), as previously described.24 Human HA-iPSCs were generated for 3 patients (1 with genotype F8 c.6429+1G>A; and 2 with intron-22 inversion, type 1) via nonintegrating episomal expression of selected reprogramming factors (OCT4, SOX2, KLF4, L-MYC, and LIN-28).25 All 3 HA-iPSC lines were tested and validated for their ability to differentiate into HA-iECs following a methodology developed by our group. Briefly, HA-iPSCs were dissociated and plated on Matrigel at a density of 40 000 cells per cm2 in mTeSR1 medium with 10 μM Y27632. After 24 hours, the medium was changed to basal medium (Advanced DMEM/F12, 1× GlutaMax, and 60 μg/mL l-ascorbic acid) supplemented with 6 μM CHIR99021. After 48 hours, cultures were dissociated into single cells and then transfected with chemically modified messenger RNA (mRNA) encoding ETS variant transcription factor 2 (ETV2; modRNA:ETV2) (TriLink BioTechnologies, LLC) by electroporation. For electroporation, 2 million cells were resuspended in 100 μL of buffer mixed with 0.8 μg of modRNA:ETV2. Electroporated cells were then seeded on a 100-mm Matrigel-coated dish in basal medium supplemented with 50 ng/mL vascular endothelial growth factor A, 50 ng/mL fibroblast growth factor 2, 10 ng/mL epidermal growth factor, and 10 μM SB431542.
PiggyBac vectors and generation of HA-FLF8-iECs
A full-length F8 gene fragment was isolated from pCDNA4/full-length FVIII (plasmid #41036; Addgene).26 This fragment was inserted into a pDONR 221 vector through BP cloning using BP Clonase II enzyme mix, then inserted into the pPB-phosphoglycerate kinase (PGK)–destination vector (plasmid #60436; Addgene) through LR cloning using LR Clonase enzyme mix.27 The final construct, PB-PGK-F8-Hyg, contains a full-length F8 open reading frame driven by a CAG promoter and a hygromycin-resistance gene driven by a PGK promoter, all flanked by 5′ and 3′ internal repeats (ITRs). A piggyBac vector containing B-domain–deleted FVIII (BDD-F8) was generated by the same method using pCDNA4/BDD-FVIII (plasmid #41035; Addgene) as the polymerase chain reaction (PCR) template.26 We picked 1 HA-iPSC line (genotype: F8 c.6429+1G>A) to insert our piggyBac with the full-length F8. To this end, HA-iPSCs were electroporated with 2.5 μg of PB-PGK-F8-Hyg piggyBac transposon vector and 0.5 μg of super piggyBac transposase expression vector (PB210PA-1; System Biosciences). Several F8-expressing HA-iPSC clones were obtained, expanded, and differentiated into HA-FLF8-iECs. Unedited HA-iECs were obtained from the same parental HA-iPSCs (genotype: F8 c.6429+1G>A) to serve as the control.
In vivo vasculogenic assay
Mice were housed in compliance with Boston Children’s Hospital guidelines, and all animal-related protocols were approved by the Institutional Animal Care and Use Committee. Immunodeficient hemophilic mice (SCID-f8ko) were developed by crossing f8ko female mice (B6;129S-F8tm1Kaz/J mice; The Jackson Laboratory) with NOD.SCID male mice (NOD.CB17-Prkdcscid/J). Vasculogenesis was evaluated in vivo using our xenograft model, as previously described.28 Grafts were histologically evaluated on day 7. Human-specific anti-CD31 antibody was used to stain human blood vessels. Perivascular cells were stained by anti-α smooth muscle actin (α-SMA) antibody. Microvessel density was reported as the average number of erythrocyte-filled vessels (vessels per mm2).
Tail-clip bleeding assay and blood plasma analysis
Mice were anesthetized, and a distal 10-mm segment of the tail was amputated with a scalpel. The tail was immediately immersed in a 50-mL Falcon tube containing isotonic saline prewarmed in a water bath to 37°C. Each animal was monitored for 20 minutes.29 Blood loss was estimated from the reduction in body weight. Upon euthanasia, blood plasma was collected from the heart in 10% sodium citrate. Plasma was then analyzed for FVIII activity using the Chromogenix Coamatic Factor VIII assay (Diapharma).
Statistical analyses
Data were expressed as mean ± standard deviation. For comparisons between 2 groups, means were compared using unpaired 2-tailed Student t tests. Comparisons between multiple groups were performed by analysis of variance (ANOVA), followed by Bonferroni posttest analysis. All statistical analyses were performed using GraphPad Prism v.5 software (GraphPad Software Inc). P < .05 was considered statistically significant.
Extended methods are described in supplemental Methods.
Results
Generation of HA-iPSCs and HA-iECs from HA patients
To generate HA-specific iPSCs, we resorted to a protocol that uses exfoliated renal epithelial cells present in urine24 (cells referred to as HA-UECs). We isolated HA-UECs from urine collected from patients with severe HA. HA-UECs were reprogramed into HA-iPSCs (Figure 1A). Subsequently, HA-iPSC colonies spontaneously emerged between days 14 and 21 (Figure 1B). Colonies were then transferred to Matrigel-coated, feeder-free culture plates for expansion. HA-iPSCs were highly pure (Figure 1D), and their phenotypes were validated by expression of pluripotent transcription factors OCT4, NANOG, and SOX2 (Figure 1C); lack of CD31 expression (Figure 1C); and by the ability to form teratomas in mice (supplemental Figure 1). Moreover, the presence of each specific patient’s genotype in the corresponding HA-iPSCs was confirmed by sequencing (supplemental Figure 2).
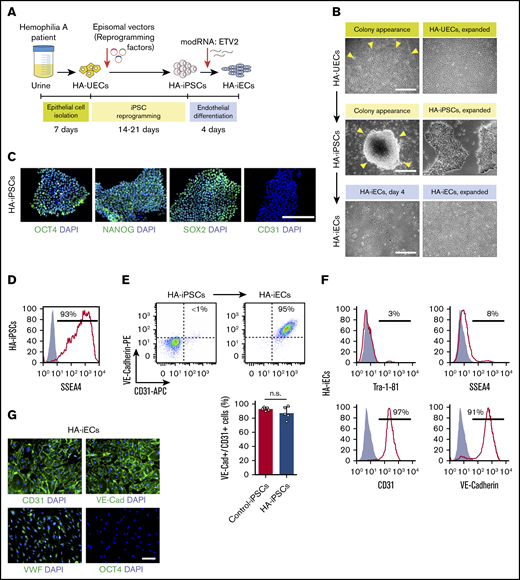
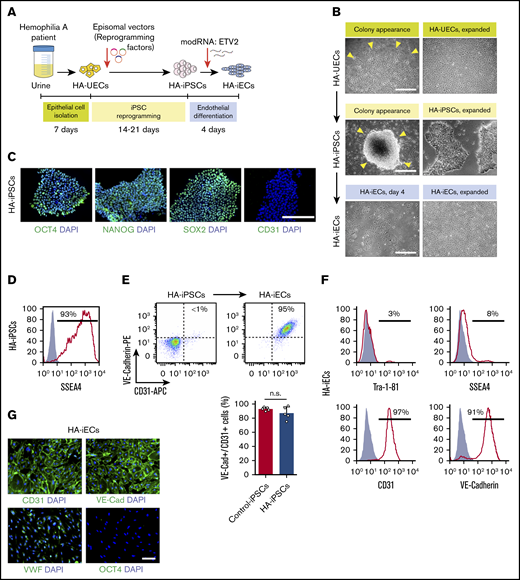
Generation of HA-iPSCs and HA-iECs from HA patients. (A) Schematic overview of epithelial cell isolation from patient urine (HA-UECs), reprogramming to HA-iPSCs through the episomal expression of reprogramming factors (OCT4, SOX2, KLF4, L-MYC, and LIN-28), and differentiation to HA-iECs using modified RNA encoding ETV2. (B) Phase-contrast imaging of the initial appearance (left) and expansion (right) of cells during the reprogramming of HA-UECs (top) to HA-iPSCs (middle), and subsequent differentiation into HA-iECs (bottom). (C) Immunofluorescence staining of HA-iPSCs for stem cell markers OCT4, SOX2, and NANOG, and endothelial cell marker CD31. Cell nuclei stained by 4′,6-diamidino-2-phenylindole (DAPI). (D) Flow cytometry analysis of HA-iPSCs for stem cell surface marker SSEA4 . Solid gray isotype-matched control is overlaid on the histogram. (E) Differentiation efficiency of HA-iPSCs into CD31+/VE-Cadherin+ HA-iECs (top right box) quantified by flow cytometry and compared with the efficiency in nonhemophilic human iPSC clones (Control-iPSCs). Bars represent mean ± standard deviation (SD). (F) Flow cytometry analysis of HA-iECs for endothelial cell surface markers CD31 and VE-Cadherin, and stem cell surface markers SSEA4 and Tra-1-81. Solid gray isotype-matched controls are overlaid on each histogram. (G) Immunofluorescent staining of HA-iECs for endothelial cell markers CD31, VE-Cadherin, and VWF, and stem cell marker OCT4. Cell nuclei stained by DAPI. Scale bars, 100 μm (G), 200 μm (C), and 500 μm (B). n.s., no statistical differences.
Generation of HA-iPSCs and HA-iECs from HA patients. (A) Schematic overview of epithelial cell isolation from patient urine (HA-UECs), reprogramming to HA-iPSCs through the episomal expression of reprogramming factors (OCT4, SOX2, KLF4, L-MYC, and LIN-28), and differentiation to HA-iECs using modified RNA encoding ETV2. (B) Phase-contrast imaging of the initial appearance (left) and expansion (right) of cells during the reprogramming of HA-UECs (top) to HA-iPSCs (middle), and subsequent differentiation into HA-iECs (bottom). (C) Immunofluorescence staining of HA-iPSCs for stem cell markers OCT4, SOX2, and NANOG, and endothelial cell marker CD31. Cell nuclei stained by 4′,6-diamidino-2-phenylindole (DAPI). (D) Flow cytometry analysis of HA-iPSCs for stem cell surface marker SSEA4 . Solid gray isotype-matched control is overlaid on the histogram. (E) Differentiation efficiency of HA-iPSCs into CD31+/VE-Cadherin+ HA-iECs (top right box) quantified by flow cytometry and compared with the efficiency in nonhemophilic human iPSC clones (Control-iPSCs). Bars represent mean ± standard deviation (SD). (F) Flow cytometry analysis of HA-iECs for endothelial cell surface markers CD31 and VE-Cadherin, and stem cell surface markers SSEA4 and Tra-1-81. Solid gray isotype-matched controls are overlaid on each histogram. (G) Immunofluorescent staining of HA-iECs for endothelial cell markers CD31, VE-Cadherin, and VWF, and stem cell marker OCT4. Cell nuclei stained by DAPI. Scale bars, 100 μm (G), 200 μm (C), and 500 μm (B). n.s., no statistical differences.
Next, we differentiated HA-iPSCs into HA-iECs. To this end, we used a 2-dimensional, feeder-free, and chemically defined protocol recently developed by our group. Briefly, HA-iPSCs were first converted into intermediate mesodermal progenitor cells via activation of Wnt signaling, a step that lasts 48 hours. Thereafter, mesodermal progenitor cells were electroporated and exposed to chemically modified RNA encoding the transcription factor ETV2 (modRNA:ETV2) (Figure 1B). This 2-step protocol rapidly and uniformly converted HA-iPSCs into HA-iECs (Figure 1B). HA-iECs uniformly expressed endothelial markers vascular endothelial (VE)–cadherin, CD31, and von Willebrand factor (VWF), and lacked expression of pluripotent markers OCT4, SSEA4, and Tra-1-81 (Figure 1E-G). Moreover, this protocol enabled a high degree of reproducibility (Figure 1E). Lastly, HA-iECs were easily expanded in culture for 2 weeks, with an average expansion yield of >40-fold (supplemental Figure 3).
Overexpression of full-length F8 in HA-iECs by piggyBac vectors
To achieve stable expression of full-length F8 in HA-iECs, we used a nonviral piggyBac DNA transposon system. The strategy was first to transfect HA-iPSCs, then select clones with high-level transgene expression of F8, and finally differentiate them into HA-FLF8-iECs (Figure 2A). Our piggyBac system was composed of 2 separate vectors. First, we constructed a 14.4-kb transposon vector with the expression of human F8 under a CAG promoter and a hygromycin-resistance gene driven by PGK between 2 ITRs for genome insertion (Figure 2A). The second vector consisted of a 7-kb plasmid encoding a super piggyBac transposase under a cytomegalovirus promoter. These 2 vectors were combined at a 5:1 ratio, respectively, and electroporated into HA-iPSCs. The resulting HA-FLF8-iPSC clones were shown to retain stem cell properties and maintain their ability to form teratomas (supplemental Figure 4). Moreover, we demonstrated that transposase activity remained negligible after HA-FLF8-iPSC cloning and after differentiation into HA-FLF8-iECs. We also showed that F8 transgene copy number remained stable after multiple passages in culture (supplemental Figure 5), and that, upon differentiation into ECs, edited HA-FLF8-iECs displayed uniform EC marker expression (supplemental Figure 6).
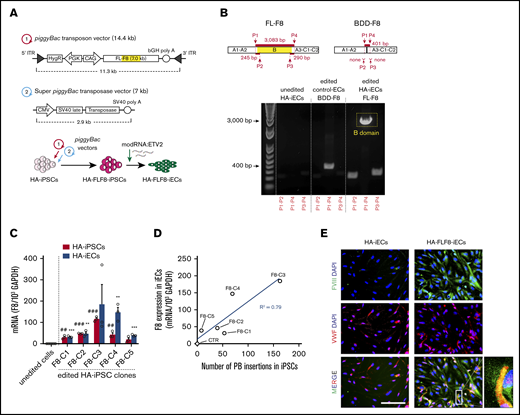
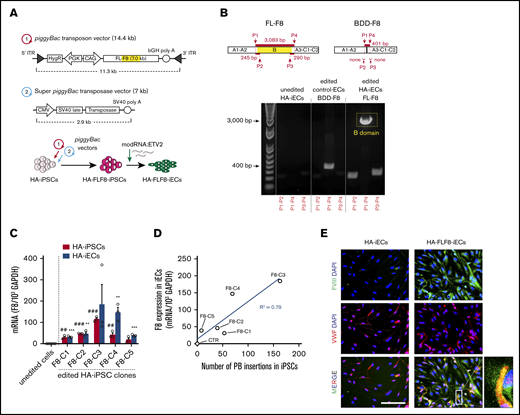
Overexpression of full-length FVIII in HA-iECs by piggyBac vectors. (A) Genetic map of (1) piggyBac transposon vector with full-length F8 (FL-F8) transgene with intact B domain (marked yellow) and (2) super piggyBac transposase expression vector. Underneath, a diagram overview of the transfection strategy of HA-iPSCs followed by differentiation into HA-FLF8-iEC. (B) Confirmation of full-length transgene insertion into HA-FLF8-iECs (right) through PCR of cDNA showing a presence of 3 fragments (∼245-bp DNA fragment for P1-P2 primers, ∼290 bp for P3-P4, and ∼3 kbp for P1-P4) compared with no endogenous bands in unedited HA-iECs (left) and a singular short fragment (∼401-bp DNA fragment for P1-P4) in control ECs that were piggyBac transfected to overexpress B-domain–deleted F8 (Control-BDD-F8-ECs; middle). (C) Quantitative reverse transcription PCR analysis confirming F8 mRNA overexpression in HA-FLF8-iPSCs and HA-FLF8-iECs in 5 independent clones (F8-C1-5). F8 expression was normalized to 103 glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Unedited HA-iPSCs and HA-iECs served as controls. Bars represent mean ± SD; ##P < .01, ###P < .001 between unedited HA-iPSCs and edited HA-FLF8-iPSC clones; **P < .01, ***P < .001 between unedited HA-iECs and edited HA-FLF8-iEC clones; n = 3. (D) Linear relationship (R2 = .79) between piggyBac (PB) insertion number (x-axis) and expression of F8 transgene (y-axis) in the 5 HA-FLF8-iEC clones and an unedited HA-iECs control. (E) Immunofluorescent costaining of FVIII (green) and VWF (red) protein in both HA-iECs and edited HA-FLF8-iECs showing overexpression of FVIII protein. Cell nuclei were stained with DAPI. Scale bar, 100 μm.
Overexpression of full-length FVIII in HA-iECs by piggyBac vectors. (A) Genetic map of (1) piggyBac transposon vector with full-length F8 (FL-F8) transgene with intact B domain (marked yellow) and (2) super piggyBac transposase expression vector. Underneath, a diagram overview of the transfection strategy of HA-iPSCs followed by differentiation into HA-FLF8-iEC. (B) Confirmation of full-length transgene insertion into HA-FLF8-iECs (right) through PCR of cDNA showing a presence of 3 fragments (∼245-bp DNA fragment for P1-P2 primers, ∼290 bp for P3-P4, and ∼3 kbp for P1-P4) compared with no endogenous bands in unedited HA-iECs (left) and a singular short fragment (∼401-bp DNA fragment for P1-P4) in control ECs that were piggyBac transfected to overexpress B-domain–deleted F8 (Control-BDD-F8-ECs; middle). (C) Quantitative reverse transcription PCR analysis confirming F8 mRNA overexpression in HA-FLF8-iPSCs and HA-FLF8-iECs in 5 independent clones (F8-C1-5). F8 expression was normalized to 103 glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Unedited HA-iPSCs and HA-iECs served as controls. Bars represent mean ± SD; ##P < .01, ###P < .001 between unedited HA-iPSCs and edited HA-FLF8-iPSC clones; **P < .01, ***P < .001 between unedited HA-iECs and edited HA-FLF8-iEC clones; n = 3. (D) Linear relationship (R2 = .79) between piggyBac (PB) insertion number (x-axis) and expression of F8 transgene (y-axis) in the 5 HA-FLF8-iEC clones and an unedited HA-iECs control. (E) Immunofluorescent costaining of FVIII (green) and VWF (red) protein in both HA-iECs and edited HA-FLF8-iECs showing overexpression of FVIII protein. Cell nuclei were stained with DAPI. Scale bar, 100 μm.
We then verified that insertion of F8 in HA-FLF8-iECs corresponded to expression of a full-length version of the gene (Figure 2B) using a combination of primers that recognize the transition between the A2 and B domains (P1-P2 primers; supplemental Table 3), and between the B and A3 domains (P3-P4) of the F8 gene (Figure 2B). Indeed, PCR analysis of cDNA from HA-FLF8-iECs revealed the presence of an ∼245-bp DNA fragment for P1-P2 primers, ∼290 bp for P3-P4, and ∼3083 bp for P1-P4, which is consistent with a full B-domain presence. In contrast, control human ECs that were piggyBac transfected with a BDD-F8 had an ∼401-bp DNA fragment for P1-P4 and lacked fragments for P1-P2 and P3-P4 (Figure 2B), as expected for a transgene lacking the B domain. It is important to note that for these experiments, we used HA-iECs derived from a cross-reacting material–positive patient (genotype: F8 c.6429+1G>A). In principle, cross-reacting material–positive patients could have positive intracellular mRNA expression for endogenous FVIII. However, we found that unedited iPSC-derived HA-iECs had virtually undetectable expression of endogenous F8 expression compared with the genetically edited ones (Figure 2B).
Next, we examined the level of F8 expression in HA-FLF8-iECs derived from 5 HA-FLF8-iPSC clones (Figure 2C-D). All clones were derived from the same HA-FLF8-iPSC line (genotype: F8 c.6429+1G>A) and displayed high-level transgene expression at the mRNA level, ranging from a 20- to 370-fold increase compared with the unedited HA-iPSC and HA-iEC controls (both from the same parental HA-iPSC line) (Figure 2C). Moreover, each of the 5 HA-iPSC clones contained multiple piggyBac insertions (ranging from 8 to 160; measured by quantitative PCR), and there was a linear correlation (R2 = 0.79) between the number of insertions detected in an HA-FLF8-iPSC clone and the level of transgene expression in the corresponding HA-FLF8-iECs (Figure 2D).
Lastly, the expression of FVIII was also corroborated in HA-FLF8-iECs at the protein level (Figure 2E). The level of FVIII expression in HA-FLF8-iECs was significantly upregulated compared with unedited HA-iECs. Of note, our immunofluorescent analysis revealed that the intracellular pattern of FVIII expression was punctuated and partially colocalized with VWF (Figure 2E), which is expected in cells that synthesize both proteins.30,31
Bioengineering HA patient-specific FVIII-secreting vascular networks in hemophilic mice
Next, we investigated whether HA-FLF8-iECs can produce functional FVIII in vivo. To this end, we first examined the capacity of HA-FLF8-iECs to engraft by forming new perfused blood vessels in hemophilic mice. We prepared grafts by mixing a suspension of HA-FLF8-iECs (derived from clone F8-C4 in Figure 2C-D; parental HA-iPSC line genotype: F8 c.6429+1G>A) and human mesenchymal stem cells (MSCs) (2 × 106 cells; 2:3 ratio, respectively) in a collagen/fibrin hydrogel as previously described.32,33 Implants containing unedited patient-derived HA-iECs served as controls. The hydrogel-cell mixtures were then subcutaneously injected into hemophilic SCID-f8ko mice, effectively creating easily identifiable and accessible implants (Figure 3A). We used SCID-f8ko mice generated in our laboratory by crossing f8ko mice with NOD.SCID mice over several generations (supplemental Figure 7). The resulting mice were genotyped, and their bleeding disorder was validated by the standard tail-tip bleeding assay.29
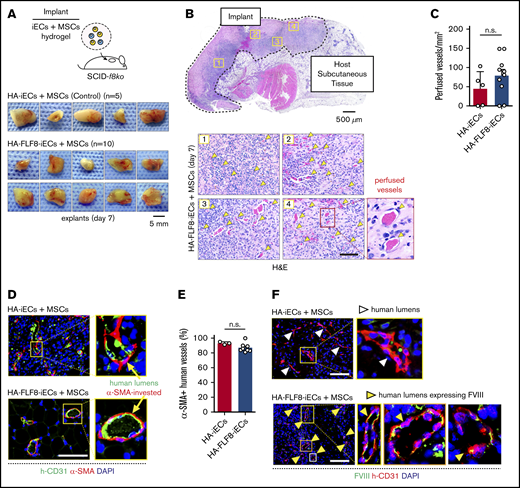
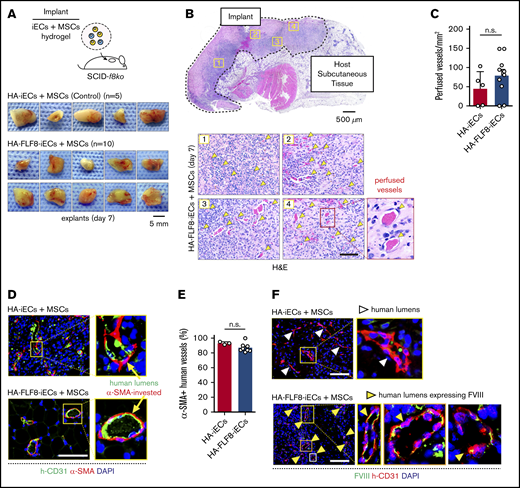
Bioengineering HA-specific FVIII-secreting vascular networks in hemophilic mice. (A) Schematic of the microvascular graft model. Grafts were prepared by combining either HA-iECs (n = 5) or HA-FLF8-iECs (n = 10) with MSCs in hydrogels followed by subcutaneous injection into immunodeficient hemophilic mice (SCID-f8ko). Images are macroscopic views of the explanted grafts at day 7. (B) Hematoxylin-and-eosin (H&E) staining of a representative explanted HA-FLF8-iEC graft. Underneath, representative pictures are taken from 4 separate regions of the graft. Perfused microvessels (yellow arrows) identified as lumenal structures containing erythrocytes. (C) Comparison of microvessel density (perfused vessels per mm2) between HA-iEC and HA-F8FL-iEC implants. Bars represent mean ± SD. (D) Immunofluorescence staining of explanted grafts after 7 days in vivo. Human lumens stained by h-CD31. Perivascular coverage stained by α-SMA. Nuclei stained by DAPI. (E) Percentage of human lumens with α-SMA+ perivascular coverage in explanted grafts on day 7. Bars represent mean ± SD. (F) Immunofluorescence staining of FVIII in explanted grafts after 7 days in vivo. Human lumens stained by h-CD31. Nuclei stained by DAPI. Human vessels in grafts formed with HA-FLF8-iECs overexpressed FVIII, whereas grafts formed by unedited HA-iECs had virtually undetectable levels of FVIII. Scale bars, 5 mm (A), 500 µm (B, gross), and 100 μm (B,D,F).
Bioengineering HA-specific FVIII-secreting vascular networks in hemophilic mice. (A) Schematic of the microvascular graft model. Grafts were prepared by combining either HA-iECs (n = 5) or HA-FLF8-iECs (n = 10) with MSCs in hydrogels followed by subcutaneous injection into immunodeficient hemophilic mice (SCID-f8ko). Images are macroscopic views of the explanted grafts at day 7. (B) Hematoxylin-and-eosin (H&E) staining of a representative explanted HA-FLF8-iEC graft. Underneath, representative pictures are taken from 4 separate regions of the graft. Perfused microvessels (yellow arrows) identified as lumenal structures containing erythrocytes. (C) Comparison of microvessel density (perfused vessels per mm2) between HA-iEC and HA-F8FL-iEC implants. Bars represent mean ± SD. (D) Immunofluorescence staining of explanted grafts after 7 days in vivo. Human lumens stained by h-CD31. Perivascular coverage stained by α-SMA. Nuclei stained by DAPI. (E) Percentage of human lumens with α-SMA+ perivascular coverage in explanted grafts on day 7. Bars represent mean ± SD. (F) Immunofluorescence staining of FVIII in explanted grafts after 7 days in vivo. Human lumens stained by h-CD31. Nuclei stained by DAPI. Human vessels in grafts formed with HA-FLF8-iECs overexpressed FVIII, whereas grafts formed by unedited HA-iECs had virtually undetectable levels of FVIII. Scale bars, 5 mm (A), 500 µm (B, gross), and 100 μm (B,D,F).
We examined our implants after 7 days in vivo. Macroscopic observation of the explants suggested similarities in the degree of vascularization between implants containing HA-FLF8-iECs (n = 10) or unedited HA-iECs (n = 5) (Figure 3A). H&E analysis revealed that grafts from both groups had extensive networks of perfused microvessels (Figure 3B), with similar microvessel densities (Figure 3C). These microvessels were primarily lined by the implanted iECs, as confirmed by the expression of human-specific CD31 (Figure 3D). Moreover, these human lumens contained mouse erythrocytes, indicating perfusion and thus the formation of functional anastomoses with the host circulatory system. Immunohistological analyses revealed that the large majority (∼80% to 100%) of the human vessels in grafts from both groups had proper coverage by perivascular cells expressing α-SMA (Figure 3D-E), a hallmark of proper vessel maturation and stabilization.34
Importantly, HA-FLF8-iECs maintained the expression of FVIII upon engraftment. Indeed, the expression of FVIII was significantly different between implants containing HA-FLF8-iECs or unedited HA-iECs. In grafts formed with HA-FLF8-iECs, human microvessels displayed a noticeable expression of FVIII at their lumens (Figure 3F). In contrast, the expression of FVIII in human microvessels lined by the unedited HA-iECs was virtually undetectable. Collectively, these results show that the genetically engineered HA-FLF8-iECs were able to engraft in the form of functional, perfused vascular networks and that they retained their ability to overexpress FVIII in vivo.
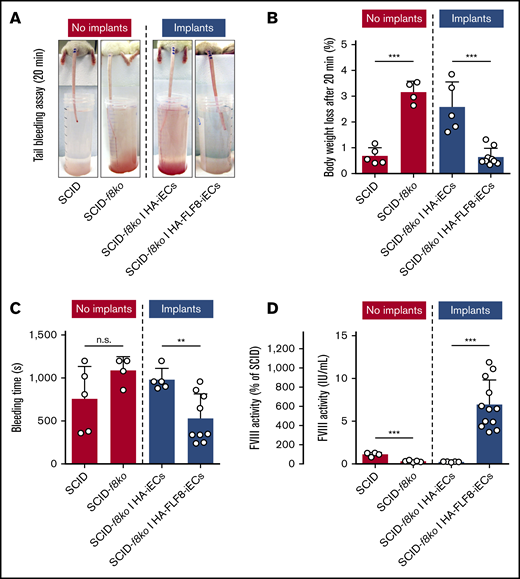
Secretion of FVIII into the bloodstream and correction of coagulation deficiency in hemophilic mice
We next sought to determine whether the newly formed microvessels lined by the HA-FLF8-iECs were able to effectively release functional FVIII into the bloodstream of the implant-bearing mice and whether the amount released was sufficient to correct their bleeding disorder. To address these questions, we subjected each implant-bearing mouse to a standardized tail-bleeding assay to assess bleeding and coagulation29 (Figure 4A). Implants containing HA-FLF8-iECs for 7 days were able to correct the clotting deficiency of the hemophilic animals and significantly decrease both percentage of body weight change and bleeding time compared with the unedited HA-iEC implant mice (Figure 4). Of note, healthy SCID mice with no implants and SCID-f8ko mice with HA-FLF8-iEC implants had similarly low body weight loss and bleeding times during the bleeding assay (Figure 4B-C). To further quantify the presence of the transgenic FVIII released by our implants, we collected blood plasma from all implant-bearing mice on day 7. Blood plasma was then analyzed for FVIII activity using the Chromogenix Coamatic Factor VIII assay with recombinant BBD-FVIII (Kogenate) as a standard control. In mice containing HA-FLF8-iEC implants, there was a significant increase in FVIII activity that was, on average, 6 times higher (∼6 IU/mL) than that of the healthy SCID controls (∼1 IU/mL). This ∼600% increase in levels of active circulating FVIII in implant-bearing mice compared with healthy mice suggested a highly efficient release of protein from our implants (Figure 4D). Of note, the capacity to secrete functional FVIII by HA-FLF8-iECs was only observed in vivo. In contrast, in vitro, we did not find differences in FVIII secretion between the edited HA-FLF8-iECs and the unedited HA-iECs (supplemental Figure 8), which suggested the importance of having HA-FLF8-iECs assembled in a proper blood vessel configuration.
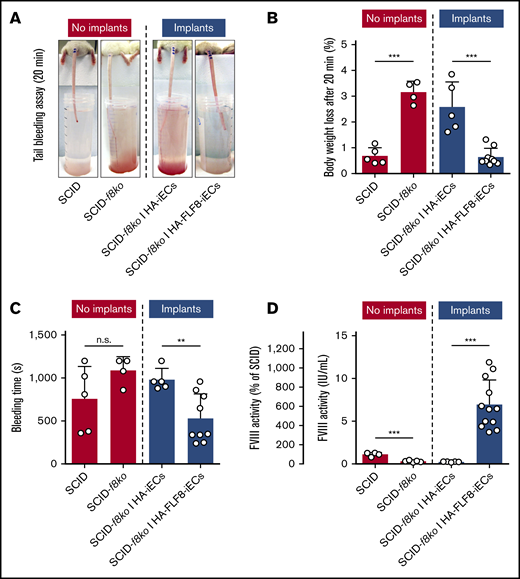
Secretion of FVIII into the bloodstream and correction of coagulation deficiency in hemophilic mice. Grafts were prepared by combining either HA-iECs (n = 5) or HA-FLF8-iECs (n = 9-12) with MSCs in collagen hydrogels followed by subcutaneous injection into immunodeficient hemophilic mice (SCID-f8ko). Tail-tip bleeding assays were performed on day 7. SCID-f8ko and nonhemophilic SCID mice with no implants served as controls. (A) Representative images of the tails after 20 minutes of the bleeding assay for each group. Percentage of body weight loss, used to quantify blood loss (B), and bleeding time (C) were recorded over the duration of the 20-minute bleeding assay with significant lowering of bleeding time and body weight loss percentage to healthy levels in mice with implants containing HA-FLF8-iECs. (D) FVIII activity levels in blood plasma collected on day 7. Levels of circulating FVIII were significantly increased in mice with implants containing HA-FLF8-iECs. Bars represent mean ± SD; **P < .01, ***P < .001.
Secretion of FVIII into the bloodstream and correction of coagulation deficiency in hemophilic mice. Grafts were prepared by combining either HA-iECs (n = 5) or HA-FLF8-iECs (n = 9-12) with MSCs in collagen hydrogels followed by subcutaneous injection into immunodeficient hemophilic mice (SCID-f8ko). Tail-tip bleeding assays were performed on day 7. SCID-f8ko and nonhemophilic SCID mice with no implants served as controls. (A) Representative images of the tails after 20 minutes of the bleeding assay for each group. Percentage of body weight loss, used to quantify blood loss (B), and bleeding time (C) were recorded over the duration of the 20-minute bleeding assay with significant lowering of bleeding time and body weight loss percentage to healthy levels in mice with implants containing HA-FLF8-iECs. (D) FVIII activity levels in blood plasma collected on day 7. Levels of circulating FVIII were significantly increased in mice with implants containing HA-FLF8-iECs. Bars represent mean ± SD; **P < .01, ***P < .001.
Our in vivo studies were originally designed to simply examine whether vessels lined by the HA-FLF8-iECs could produce and secrete full-length FVIII into the bloodstream. Thus, we used the very simple subcutaneous xenograft model lasting only 7 days. Nevertheless, it is important to note that this mode of cell engraftment into the subcutaneous space usually results in human ECs being rapidly replaced by the host murine ECs, a turnover that is not ideal for long-term studies. Indeed, the examination of our grafts at 4 weeks revealed that the initial abundant presence of human vessels (lined by hC31+/mCD31− cells) at 1 week was replaced by a majority of vessels lined by host murine ECs (hCD31−/mCD31+ cells) at 4 weeks (supplemental Figure 9A). Consequently, although all grafts were deemed successful at 1 week (n = 10 of 10), the rate of success was progressively (and significantly) reduced at 2 weeks (only 3 of 7) and 4 weeks (1 of 6) (supplemental Figure 9B). These results highlighted the importance of conducting further studies to assess durability by transplanting grafts at sites that are more favorable to long-lasting EC engraftment than the subcutaneous space.
In summary, our results show significant restoration of hemostasis (albeit only at 1 week) and validate the proof of concept that our genetically engineered patients’ cells (HA-FLF8-iECs) can be engrafted in the form of microvessels within a confined implant. These microvessels can, in turn, produce and secrete functional FVIII into the bloodstream of the animals, restoring therapeutic levels of FVIII activity and treating HA.
Discussion
Here, we have developed a nonviral approach to genetically engineer HA patient-specific ECs for full-length FVIII overexpression. We demonstrated that the modified HA-FLF8-iECs could form FVIII-secreting microvascular networks upon implantation in vivo. The concept was to bioengineer a microvascular graft in which the endothelium is lined by patients’ cells that were genetically engineered to carry out a drug delivery role.33 Importantly, our bioengineered vascular networks are confined inside implants that remain easily accessible and thus retrievable.
Over the last few decades, HA has been a particularly appealing target for gene therapy.15 Currently, this is predominantly done with viral-based approaches. One of the motivations of our study, however, was to avoid using viruses and to circumvent the limitation imposed by a restricted viral cargo size. We sought to establish an alternative approach that allows transfecting patients’ cells with the full-length version of the F8 gene (∼7.0-kb cDNA). To this end, we implemented a nonviral piggyBac DNA transposon strategy that can insert large genetic cargos.35 Indeed, we were able to encode for the full-length version of the human F8 gene and inserted multiple copies of it into the target cells (ranging from 8 to 160), a notable advantage of the piggyBac system.36 Of note, all genetic modifications were performed at the iPSC level, allowing us to easily select for clones of HA-FLF8-iPSC with high levels of F8 expression. Although we did not optimize the system for maximal insertion, future studies could investigate a potential ceiling for the number of F8 inserts before the loss of cell function or cytotoxicity may occur. Collectively, the use of the piggyBac transposon system was extremely instrumental for our study. Previously, preclinical research by Matsui et al used a piggyBac vector that encoded full-length F8 in a murine model of in vivo gene therapy.35 However, to our knowledge, our study is the first to use the piggyBac system in the context of ex vivo gene therapy, and that resulted in successful overexpression and secretion of full-length FVIII.
As a central part of our strategy, we resorted to genetically engineering HA patients’ ECs ex vivo. In principle, ex vivo gene therapy guarantees specificity because cells are genetically modified in vitro under controlled conditions. The differentiation of HA-iPSCs into HA-iECs was carried out with high efficiency, independently of the HA-iPSC clone selected. Our usage of HA-iECs was deliberate, and the reasons twofold: (1) ECs are the natural producers of FVIII in the body.37 Also, if FVIII expression is induced in a cell that synthesizes and stores VWF, FVIII will interact with VWF and be stored and released as a noncovalent complex.31 We showed that upon transfection, overexpressed FVIII partially colocalized with VWF in the modified HA-FLF8-iECs (Figure 2E). (2) ECs will inherently line the lumen of the vasculature upon engraftment and therefore will secrete FVIII directly into the bloodstream. Indeed, in our subcutaneous grafts, the presence of lumenal structures that overexpressed human FVIII was evident, as well as the high levels of active FVIII in the plasma of implant-bearing hemophilic mice (Figures 3F and 4D). We demonstrated that HA-FLF8-iECs were able to self-assemble into a microvascular network that formed anastomoses and connected with the host circulatory system. This mode of engraftment, in turn, allowed the implanted ECs to rapidly adopt a proper physiological role, lining the lumen of perfused vessels, which facilitates integration with the host. Certainly, alternative modes of EC engraftment have been proposed. For example, Xu et al, injected normal (nonhemophilic) murine iPSC-derived ECs into the liver of hemophilic mice, correcting their bleeding deficiency.38 Olgasi et al engrafted human iPSC-derived, FVIII-expressing ECs into the liver of nonobese diabetic (NOD) SCID-IL2rγ null hemophilic mice, effectively restoring hemostasis.39 However, notwithstanding successful efficacy, this approach of disseminating cells in the liver complicates future accessibility and reversibility. In contrast, our mode of engraftment results in tight cellular confinement inside excisable implants, which should allow more effective monitoring and reversibility.33
Other ex vivo gene-therapy studies have previously used ECs and are of significant importance in the field.39,40 The majority of these studies, however, relied on viral vectors, and none produced full-length FVIII. Editing the defective endogenous F8 gene in patients’ ECs has also been proposed.41 However, it remains unclear whether simply correcting the endogenous F8 gene will be able to yield therapeutic levels of FVIII when scaling up. Studies in mice have shown that engrafting human ECs that were either genetically edited (ie, endogenous F8 corrected)41 or lentivirally engineered with low numbers of a BDD-F8 transgene39,42,43 restored FVIII activity to only ∼2% to 11% of normal phenotypic levels. Alternatively, we inserted up to 160 new copies of full-length F8 cDNA with our piggyBac approach, allowing us to raise circulating FVIII activity in hemophilic mice up to ∼600% of the level of a control SCID mouse. Using ECs genetically engineered to contain multiple copies of an overexpressed transgenic full-length F8 gene may offer advantages in scalability. Also, it is important to note that our piggyBac system entails the expression of F8 under a strong synthetic CAG promoter. The selection of the CAG promoter was intended to assure sufficiently high expression of FVIII in grafts containing a limited number of ECs. Nevertheless, other studies have successfully demonstrated the use of EC-specific promoters, including the native F8 promoter, to direct gene replacement to FVIII-secreting cells.39,44
Previous ex vivo gene-therapy studies have also included the use of alternative nonendothelial cells as therapeutic FVIII delivery vehicles, including hematopoietic stem cells (HSCs).19-21 This approach is particularly appealing due to the feasibility of long-term HSC engraftment, the ability for these HSCs to permanently generate FVIII-containing platelets, and the potential to avoid inhibitors to FVIII in patients due to platelet sequestering. However, drawbacks of this approach include the need for radiation of the host (or busulfan treatment) to enable HSC engraftment, and the intrinsic inaccessibility and thus irreversibility of the treatment (ie, no confined implant that can be easily retrieved).
It is important to note that our study is simply an initial proof of concept, and, as such, it has several limitations. A first limitation is the lack of insights regarding the immune response. One of the theoretical advantages of using a nonviral piggyBac DNA transposon system is that this should eliminate the risk of potential adverse immunological reactions against viral proteins.45,46 In addition, some viral vectors, like those based on AAV, might be ineffective for patients who have been previously exposed to wild-type viruses due to the development of neutralizing antibodies.47 Nevertheless, our study was conducted in immunodeficient (SCID background) mice; thus, we were not able to examine immune-related questions, including whether the use of autologous cells would avoid an immune response altogether. To adequately address these questions, future studies should be conducted using humanized mouse models that account, to some extent, for the effects produced by the immune system. Also, additional future studies should be carried out in an autologous setting, using syngeneic immunocompetent large animal models of HA (ie, a dog model).
A significant focus of our study was to demonstrate that it is feasible to overexpress the full-length version of the F8 gene in HA patient-derived cells. However, we did not systematically compare, side-by-side, the potential therapeutic advantages of full-length FVIII with those of B-domain–deleted FVIII (BDD-FVIII). Currently, the use of BDD-FVIII is prominent in HA gene therapy as it circumvents the inability of most viral vectors to carry the full-length F8 gene. Nonetheless, questions about the implications of using a truncated BDD-FVIII instead of the full-length version remain. On the one hand, previous studies in the setting of recombinant protein expression have demonstrated that using BDD-FVIII provides a significant increase in secretion over FL-FVIII.48 On the other hand, however, evidence mounts with regard to the role of the B domain in critical functions such as FVIII secretion into the bloodstream and clearance from plasma.16-18,35 Thus, future studies are warranted to compare, side-by-side, the performance of BDD-FVIII vs FL-FVIII in the context of our ex vivo gene-therapy approach.
A third limitation of the study is that we did not assess durability and long-term efficacy. Our study was designed to examine only initial feasibility, with the hemophilic mice carrying the implants for only 1 week. Also, it is important to note that when human vessels are implanted subcutaneously in a mouse, there is a natural turnover of the endothelium that results in the human ECs being progressively replaced by the host murine ECs, which, in turn, affect the ability to produce FVIII in our grafts (supplemental Figure 9). This limitation is intrinsic to the subcutaneous xenograft model and precluded properly examining durability. Certainly, the clinical feasibility of our approach would depend on achieving long-term durability, and thus further studies are warranted. These future studies should be ideally conducted by transplanting grafts at sites that are more favorable to long-lasting EC engraftment than the subcutaneous space. For example, previous studies have shown that human iPSC-derived ECs that were intraportally transplanted into hemophilic SCID mice were still present in the liver after 12 weeks.39 The renal capsule is another site that has been proven to be reasonably stable for microvascular engraftment.49 In addition, to properly address the question of human graft durability, the immunodeficient murine model might not be ideal. Alternatively, future studies should be conducted using humanized mouse models that account for the effect produced by the immune system on the durability of the grafts. In any case, the success of our approach undoubtedly depends on achieving the long-term durability of the vessels, and this requires further investigations. These future investigations should not only evaluate efficacy but also estimate the number of iECs effectively remaining in the grafts over time.
In summary, our studies established the feasibility of a nonviral approach to genetically engineer HA patient-specific ECs for full-length FVIII overexpression. Moreover, we demonstrated that the modified HA-FLF8-iECs could form FVIII-secreting vascular networks within subcutaneous implants in hemophilic mice, restoring therapeutic levels of FVIII activity. We envision that this proof-of-concept study could become the basis for a new autologous ex vivo gene-therapy approach to treat HA. Nevertheless, further investigations are warranted to determine the effects of the immune system, the advantages and disadvantages of expressing FL-FVIII vs BDD-FVIII in HA-iECs, and to establish the potential for long-term durability of this approach.
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01AR069038 [National Institute of Arthritis and Musculoskeletal and Skin Diseases], R01HL128452 [National Heart, Lung, and Blood Institute], and R21AI123883 [National Institute of Allergy and Infectious Diseases]) and from the Bayer Hemophilia Awards Program (J.M.M.-M). Histology was supported by the Core Facility of the Dana-Farber/Harvard Cancer Center (National Cancer Institute grant P30 CA06516). Kogenate FS, used as the standard in the Chromogenix Coamatic FVIII assay, was supplied by Bayer Healthcare Pharmaceuticals Inc.
Authorship
Contribution: J.N., R.-Z.L., and J.M.M.-M. conceived and designed the project; J.N., R.-Z.L., X.H., and K.W. performed experimental work; all authors discussed and analyzed the data and edited the results; S.E.C., T.H., and E.J.N. provided crucial material; and J.N. and J.M.M.-M. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for E.J.N. is St. Jude Children’s Research Hospital, Memphis, TN.
Correspondence: Juan M. Melero-Martin, Department of Cardiac Surgery, Boston Children’s Hospital, 300 Longwood Ave, Boston, MA 02115; e-mail: juan.meleromartin@childrens.harvard.edu.