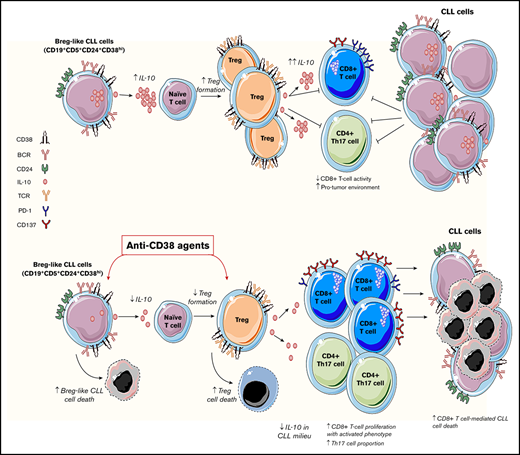
Key Points
CD38hi Breg-like CLL cells promote conversion of naive T cells into Tregs in an IL-10/TGF-β–dependent manner.
CD38 targeting drugs are lethal to both IL-10–producing Breg-like CLL cells and Tregs, but lead to improved CD8+ T-cell cytolytic responses.

Abstract
Patients with chronic lymphocytic leukemia (CLL) are characterized by monoclonal expansion of CD5+CD23+CD27+CD19+κ/λ+ B lymphocytes and are clinically noted to have profound immune suppression. In these patients, it has been recently shown that a subset of B cells possesses regulatory functions and secretes high levels of interleukin 10 (IL-10). Our investigation identified that CLL cells with a CD19+CD24+CD38hi immunophenotype (B regulatory cell [Breg]–like CLL cells) produce high amounts of IL-10 and transforming growth factor β (TGF-β) and are capable of transforming naive T helper cells into CD4+CD25+FoxP3+ T regulatory cells (Tregs) in an IL-10/TGF-β-dependent manner. A strong correlation between the percentage of CD38+ CLL cells and Tregs was observed. CD38hi Tregs comprised more than 50% of Tregs in peripheral blood mononuclear cells (PBMCs) in patients with CLL. Anti-CD38 targeting agents resulted in lethality of both Breg-like CLL and Treg cells via apoptosis. Ex vivo, use of anti-CD38 monoclonal antibody (mAb) therapy was associated with a reduction in IL-10 and CLL patient-derived Tregs, but an increase in interferon-γ and proliferation of cytotoxic CD8+ T cells with an activated phenotype, which showed an improved ability to lyse patient-autologous CLL cells. Finally, effects of anti-CD38 mAb therapy were validated in a CLL–patient-derived xenograft model in vivo, which showed decreased percentage of Bregs, Tregs, and PD1+CD38hiCD8+ T cells, but increased Th17 and CD8+ T cells (vs vehicle). Altogether, our results demonstrate that targeting CD38 in CLL can modulate the tumor microenvironment; skewing T-cell populations from an immunosuppressive to immune-reactive milieu, thus promoting immune reconstitution for enhanced anti-CLL response.
Introduction
Chronic lymphocytic leukemia (CLL) is a B-cell cancer in which there is a concomitant dysregulation of the nonmalignant T-cell compartment and immune cytokine milieu.1-5 T cells from patients with CLL are often impaired, with a notable increase in T regulatory cells (Tregs) and compromised CD8+ cytotoxic T-cell (cTL) functionality.6,7 These cellular deviations are accompanied with dysregulation of immunosuppressive cytokines such as interleukin 10 (IL-10) and transforming growth factor β (TGF-β) secretion.2,8 Together, these changes contribute to clinical progression of disease.9
Abnormalities in the T-cell population are present at early stages of disease in patients with CLL, suggesting the ability of malignant B-CLL clones (even in low numbers) to exert a dominant effect on their microenvironment.10,11 It has recently been shown that within the overall CLL cell compartment, a subset of B-CLL cells phenotypically resemble and function as B regulatory cells (Bregs).12 Bregs constitute a newly designated group of B cells that have the capability to exert suppressive effects on a variety of immune cell types,13 mediated in part by IL-10 secretion. Several types of Bregs have been identified,14 with 1 subset having a CD19+CD24+CD38hi immunophenotype and an enhanced capacity to secrete IL-10 (termed B10 Bregs). B10 Bregs are highly immunosuppressive and can dampen effector CD4+ T helper cells, specifically Th1 and Th17 cell responses,15,16 and also impair cTL activity.
Patients with CLL with at least 30% CD38+ B-CLL cells are designated as having CD38+ disease, which is associated with an unfavorable clinical prognosis.17 We have recently demonstrated that targeting CD38 with the anti-CD38 human monoclonal antibody (mAb) daratumumab downregulates B-cell receptor signaling and enhances the antitumor activity of ibrutinib in CLL cells and Waldenstrom macroglobulinemia tumor cells.18,19 These investigations revealed that daratumumab induces antibody-dependent cell-mediated cytotoxicity, complement-dependent cytotoxicity, antibody-dependent cellular phagocytosis, and direct apoptosis of CLL cells in vitro, and together these undergird its anti-CLL activity in vivo.
As CLL cells are highly dependent on their interaction with neighboring immune cells,20,21 we investigated the effects of CD38-targeting agents on Breg-like CLL cells, T-cell subsets, their associated immune cytokine environment, and the downstream effect on functionality of cTLs from patients with CLL.
Materials and methods
Human samples, T-cell assays, mouse model, and statistical analysis
Peripheral blood mononuclear cells (PBMCs) from patients with CLL (n = 22 with ≥90% tumor B cells; clinical/biological data in Table 1) and healthy donors were isolated under a protocol approved by the Mayo Clinic Institutional Review Board. CD19+CD5+CD38hi/lo CLL cells and CD4+CD25+CD127lo Tregs were sorted out using magnetic beads/flow-sorter (sorting and gating strategy in supplemental Materials and methods).18 A CLL–patient-derived xenograft (PDX) model was established,22 using PBMCs isolated from a patient with CLL with CD38+ disease, injected into NSG mice (The Jackson Laboratory). iTreg formation assays were carried out using naive Th cells prestimulated with anti-CD3 (5 µg/mL)/CD28 (5 µg/mL) antibodies followed by coculture with either autologous Breg-like (CD19+CD24+CD38+) or non-Breg (CD19+CD24+CD38−) CLL cells. cTL proliferation was determined using CellTrace carboxyfluorescein succinimidyl ester (Thermo Scientific) on a flow cytometer. cTL cytolytic activity was measured by co-culturing cTLs with calcein-AM–labeled autologous/allogeneic CLL cells for 6 hours. For experiments in which CD38 expression was assessed in cells treated with daratumumab or kuromanin, a multiepitope fluorescein isothiocyanate-conjugated anti-CD38 antibody (Cytognos, Salamanca, Spain) was used as previously described by us.18 Daratumumab was acquired through clinical sources; small molecule CD38 inhibitors, kuromanin23 and 78c,24,25 were purchased from Selleckem and provided as a gift from Eduardo Chini (Mayo Clinic, Rochester, MN), respectively. All resulting values are expressed as mean ± standard error of the mean (SEM). Data were analyzed for significance in GraphPad Prism by paired Student t test, Mann-Whitney U test, or 1-way analysis of variance with Bonferroni adjustment as specified. Correlation coefficients and their significance were calculated by 2-tailed Spearman’s rank correlation. A confidence interval of 95% or P < .05 was considered statistically significant.
Detailed descriptions of the materials and methods are provided in the supplemental Materials and methods.
Results
CD38-targeting agents decrease IL-10 and increase IFN-γ in the CLL-immune milieu
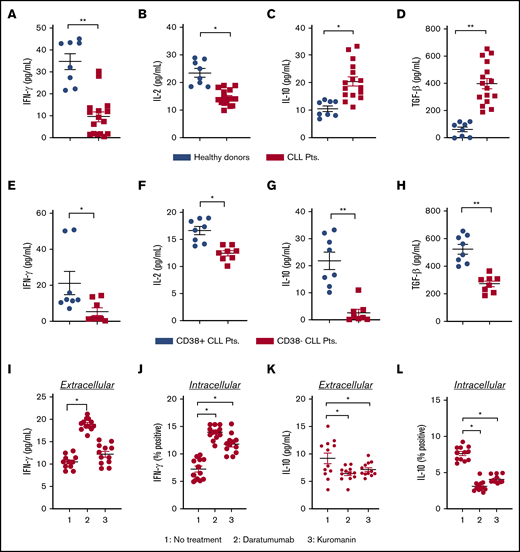
The immune system of patients with CLL is highly dysfunctional and poised toward an immunosuppressive disposition, which leads to compromised antitumor immune-effector activity and poor clinical outcome.26 Increased IL-10 and TGF-β (anti-inflammatory), along with decreased IL-2 and interferon-γ (IFN-γ) (proinflammatory), are known to promote tumor growth and dampen immune functionality.12,13 Cytokine analysis from the plasma of patients with CLL revealed IFN-γ and IL-2 levels to be 3.6- and 1.5-fold lower vs healthy donors (P = .0022 and P = .0065, respectively; Figure 1A-B). Conversely, plasma levels of IL-10 and TGF-β were two- and 6.4-fold higher in patients with CLL vs healthy donors (P = .001 and P = .0012, respectively; Figure 1C-D). As increased expression of CD38 on leukemic cells is a well-established parameter that confers poor clinical prognosis,17 we performed subset analysis of cytokine levels in CD38+ or CD38− patients with CLL. Expression of IFN-γ (Figure 1E; P = .0281), IL-2 (Figure 1F; P = .0006), IL-10 (Figure 1G; P = .0003), and TGF-β (Figure 1E; P = .0002) in the plasma of CD38+ patients was significantly higher than in CD38− patients. It has been previously reported that CD38 activation can increase IL-10 levels in monocytes.27 Conversely, inhibition of cADPR, a substrate that is biosynthesized by CD38 NADase activity, results in decreased expression of TGF-β.28 We hypothesized that targeting CD38 on CLL cells would decrease IL-10 and TGF-β while raising IFN-γ and IL-2 levels. Exposure of PBMCs from patients with CLL to daratumumab raised the extracellular concentration of IFN-γ (Figure 1I; P = .022), as well as intracellular expression of IL-10, in gated CLL cells (Figure 1J; P = .0286) by nearly twofold compared with untreated cells. With kuromanin, a significant increase in intracellular IFN-γ was noted in gated CLL cells (P = .018); however, this was not evident in the extracellular fraction. Examination of extracellular IL-10 in daratumumab-treated (P = .0274) or kuromanin-treated PBMCs from patients with CLL (P = .032) was significantly lower than in their untreated counterparts (Figure 1K). A similar decrease in intracellular IL-10 (and IL-10 mean fluorescence intensity [MFI]; supplemental Figure 1) was noted in the CD19+ gated fraction of daratumumab (P = .0125) or kuromanin-treated PBMCs (P = .0158; Figure 1L). Altogether, these results demonstrate that targeting CD38 (by either therapeutic mAb or small molecule) can alter cytokines (IL-10, IFN-γ) involved in regulating the interaction between CLL cells and neighboring immune cells.
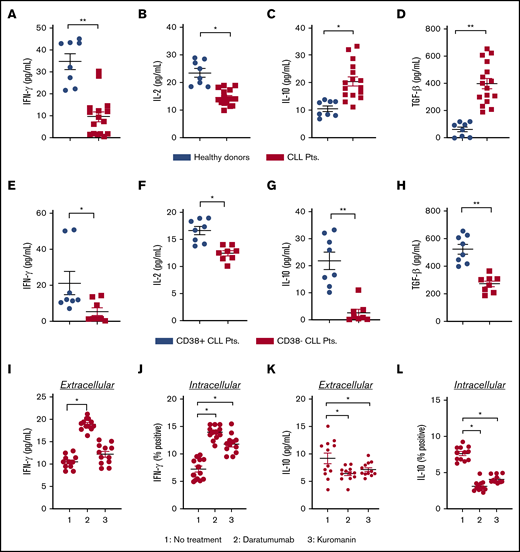
Effect of anti-CD38 agents on the level of pro and anti-inflammatory cytokines in patients. (A-D) Plasma from healthy donors (n = 8) or patients with CLL (n = 16) were isolated from blood, and concentration of IFN-γ (A) and IL-2 (B) was measured by sandwich enzyme-linked immunosorbent assay. Similarly, levels of IL-10 (C) and TGF-β (D) were assessed. From the overall data set, subset analysis of IFN-γ (E), IL-2 (F), IL-10 (G), and TGF-β (H) levels in patients with clinically defined CD38+ (n = 8) and CD38− (n = 8) disease was conducted. (I-J) The effect of daratumumab (1 µg/mL, 72 hours) or kuromanin (30 µM, 72 hours) on extracellular (I) IFN-γ from patients with CLL was examined (n = 12, n = 3 with CD38− disease and n = 9 with CD38+ disease). After drug treatment with daratumumab or kuromanin, supernatant from the PBMCs of patients with CLL was collected via centrifugation and cytokines measured by sandwich enzyme-linked immunosorbent assay. (J) Intracellular IFN-γ positivity in CD19+ gated CLL cells (from the same patients as in panel I) was measured after treatment with daratumumab or kuromanin (same concentrations/time as in panel I); 4 hours before termination of experiments, ion channels were blocked with ionomycin and brefeldin used for GolgiStop. Extracellular IL-10 (K) and IL-10+ (L) CLL cells were measured in the same manner as in panels I and J, respectively. For extracellular cytokines, data were derived from a standard curve and represented as picograms per milliliter ± SEM in a scatter dot plot for each set of experiments. For intracellular staining, data were represented as percentage positive cells ± SEM. Each experiment was performed at least thrice in duplicate. *P < .05; **P < .001.
Effect of anti-CD38 agents on the level of pro and anti-inflammatory cytokines in patients. (A-D) Plasma from healthy donors (n = 8) or patients with CLL (n = 16) were isolated from blood, and concentration of IFN-γ (A) and IL-2 (B) was measured by sandwich enzyme-linked immunosorbent assay. Similarly, levels of IL-10 (C) and TGF-β (D) were assessed. From the overall data set, subset analysis of IFN-γ (E), IL-2 (F), IL-10 (G), and TGF-β (H) levels in patients with clinically defined CD38+ (n = 8) and CD38− (n = 8) disease was conducted. (I-J) The effect of daratumumab (1 µg/mL, 72 hours) or kuromanin (30 µM, 72 hours) on extracellular (I) IFN-γ from patients with CLL was examined (n = 12, n = 3 with CD38− disease and n = 9 with CD38+ disease). After drug treatment with daratumumab or kuromanin, supernatant from the PBMCs of patients with CLL was collected via centrifugation and cytokines measured by sandwich enzyme-linked immunosorbent assay. (J) Intracellular IFN-γ positivity in CD19+ gated CLL cells (from the same patients as in panel I) was measured after treatment with daratumumab or kuromanin (same concentrations/time as in panel I); 4 hours before termination of experiments, ion channels were blocked with ionomycin and brefeldin used for GolgiStop. Extracellular IL-10 (K) and IL-10+ (L) CLL cells were measured in the same manner as in panels I and J, respectively. For extracellular cytokines, data were derived from a standard curve and represented as picograms per milliliter ± SEM in a scatter dot plot for each set of experiments. For intracellular staining, data were represented as percentage positive cells ± SEM. Each experiment was performed at least thrice in duplicate. *P < .05; **P < .001.
A subset of CLL cells resembles B10 Bregs, produces IL-10, and can be eradicated by CD38-targeting agents
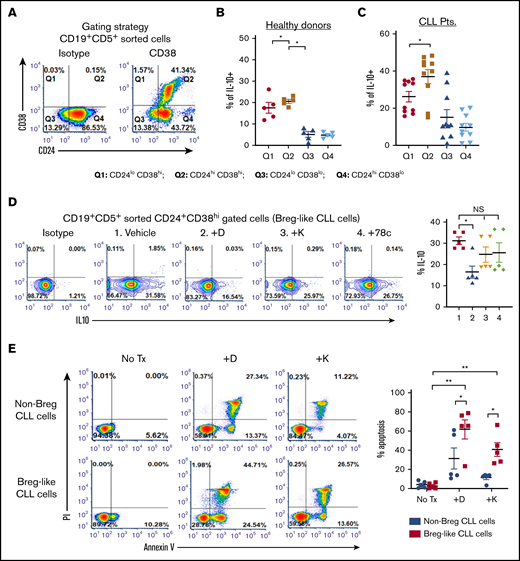
A subset of CLL cells resembles Bregs and shares IL-10 competence.12,15 We noted that IL-10 was significantly higher within the CD19+CD5+CD24+CD38hi fraction of CLL cells (Breg-like CLL cells) relative to the CD19+CD5+CD24+CD38lo fraction (non-Breg CLL cells) by nearly twofold (P = .006 and 0.0012; Figure 2A-C). We hypothesized that pharmacologic interference of CD38 would decrease the percentage of IL-10+ Breg-like CLL cells. Compared with vehicle, intracellular IL-10 in Breg-like CLL cells was significantly decreased by daratumumab (from 31.58% to 17.79%; P = .019), but not with either kuromanin or 78c (Figure 2D). In parallel experiments and in line with our recently published data,18 we noted significant apoptosis of Breg-like CLL cells (Figure 2E). Taken together, we show that Breg-like CLL cells are a source of IL-10 production, but are eradicated by daratumumab, which is associated with an overall decrease in IL-10 levels.
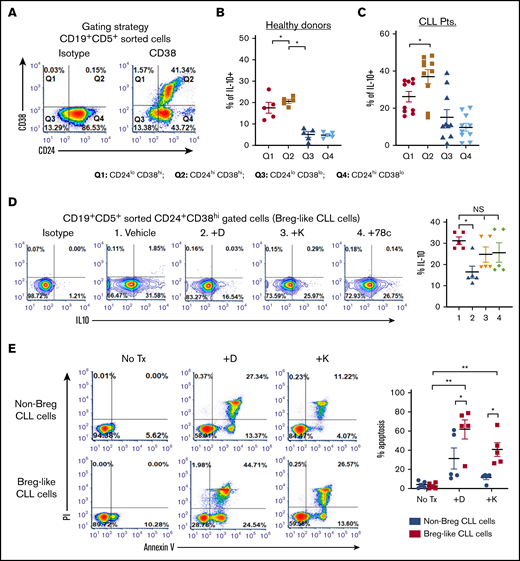
Breg-like CLL cells produce IL-10 and are highly sensitive to the cytotoxic effects of anti-CD38 agents. (A-C) PBMCs from patients with CLL (n = 10) and healthy donors (n = 5), were CD19+CD5+ flow sorted and further gated based on CD24+CD38+ surface expression (A) (representative density plot shown to illustrate gating strategy: Q1, CD24loCD38hi; Q2, CD24hiCD38hi; Q3, CD24loCD38lo; and Q4, CD24hiCD38lo) followed by measurement of intracellular IL-10 within the PBMC fraction from healthy donors (B) and patients with CLL (C). (D) PBMCs from patients with CLL (n = 5) were treated with vehicle, daratumumab (1 µg/mL), kuromanin (30 µM), or 78c (0.5 µM), followed by quantification of Breg-like CLL cells. (E) PBMCs from patients with CLL (n = 5) were sorted to enrich for Breg-like CLL cells (CD19+CD5+CD24+CD38+) and non-Breg CLL cells (CD19+CD5+CD24+CD38−) and treated with vehicle, daratumumab (1 µg/mL), or kuromanin (30 µM). After 24 hours, cells were stained with annexin V and propidium iodide (PI) and percentage apoptotic cells measured on a flow cytometer. Contour or density plots are representative and compiled data are presented as mean ± SEM, with individual data points overlaid. Each experiment was performed at least twice in duplicate. *P < .05; **P < .001. NS, not significant.
Breg-like CLL cells produce IL-10 and are highly sensitive to the cytotoxic effects of anti-CD38 agents. (A-C) PBMCs from patients with CLL (n = 10) and healthy donors (n = 5), were CD19+CD5+ flow sorted and further gated based on CD24+CD38+ surface expression (A) (representative density plot shown to illustrate gating strategy: Q1, CD24loCD38hi; Q2, CD24hiCD38hi; Q3, CD24loCD38lo; and Q4, CD24hiCD38lo) followed by measurement of intracellular IL-10 within the PBMC fraction from healthy donors (B) and patients with CLL (C). (D) PBMCs from patients with CLL (n = 5) were treated with vehicle, daratumumab (1 µg/mL), kuromanin (30 µM), or 78c (0.5 µM), followed by quantification of Breg-like CLL cells. (E) PBMCs from patients with CLL (n = 5) were sorted to enrich for Breg-like CLL cells (CD19+CD5+CD24+CD38+) and non-Breg CLL cells (CD19+CD5+CD24+CD38−) and treated with vehicle, daratumumab (1 µg/mL), or kuromanin (30 µM). After 24 hours, cells were stained with annexin V and propidium iodide (PI) and percentage apoptotic cells measured on a flow cytometer. Contour or density plots are representative and compiled data are presented as mean ± SEM, with individual data points overlaid. Each experiment was performed at least twice in duplicate. *P < .05; **P < .001. NS, not significant.
CLL cells promote conversion of naive T-helper cells into Tregs in an IL-10/TGF-β-dependent manner
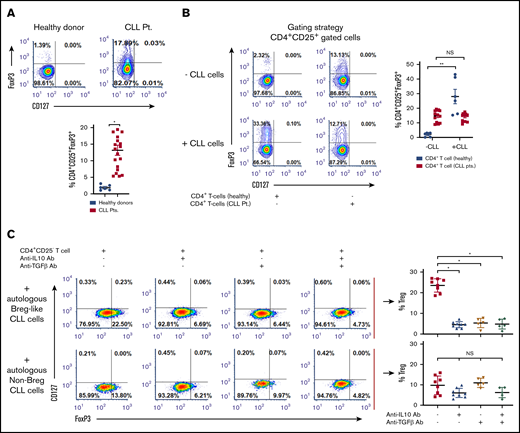
Tregs can dampen antitumor CD4+ or CD8+ T-cell responses and promote neoplastic B-cell growth.6,29 We noted a significantly higher proportion of Tregs present within PBMCs from patients with CLL vs healthy donors (11.86% ± 2.03% vs 1.94% ± 0.32%; P = .026; Figure 3A) and which was significantly higher in CD38+ (16.10% ± 2.17%) vs CD38− patients with CLL (9.92% ± 1.38%; P = .04; supplemental Figure 2A-B). Absolute cell counts and percentage Tregs assessed from 19 patients with CLL are provided in supplemental Table 1. We hypothesized that Tregs in patients with CLL may be increased because of the influence of CLL cells on naive T helper cells. In a coculture of CD19+CD5+ CLL cells with patient-autologous CD3/CD28-stimulated naive T helper cells or those independently isolated from healthy donors, we noted a 7.5-fold induction of FoxP3+ Tregs (iTregs) in the coculture containing naive T helper cells from healthy donors (P = .0014). Coculture between CLL cells and autologous naive T helper cells did not induce increased iTreg formation (Figure 3B). Next, in a transwell assay, we placed magnetically isolated CLL Bregs or, separately, non-Breg CLL cells (upper chamber) in proximity with patient-autologous naive T helper cells (lower chamber), and observed significantly more induction of iTregs (23.65% ± 1.15%) in the transwell containing CLL Bregs relative to the transwell containing non-Breg CLL cells (9.91% ± 1.61%; P = .011; Figure 3C, top and bottom red dot plots). We reasoned that increased iTreg formation may be a result of soluble IL-10 and/or TGF-β secreted mainly from Breg-like CLL cells, inferred from the proportion of IL-10+ or TGF-β+ cells being higher in CD38+ patients with CLL (Figure 1G; supplemental Figure 3). Indeed, in the transwell containing Breg-like CLL cells and naive T helper cells, an anti-IL-10 or anti-TGF-β neutralizing Ab significantly reduced iTreg formation by nearly five- or 4.3-fold, respectively (compare top red scatter dot plot points with adjacent blue and brown points; P = .0068). The combination of the 2 neutralizing antibodies did not result in a greater decrease of iTreg formation than that noted with singular use of anti-IL-10 or anti-TGF-β neutralizing Abs. Contrastingly, in the transwell with non-Breg CLL cells, insignificant reductions in iTreg formation were noted with anti-IL-10+/− anti-TGF-β neutralizing Abs (Figure 3C). These novel findings prompted us to conclude that Breg-like CLL cells are capable of promoting iTreg formation in an IL-10- and TGF-β-dependent manner.
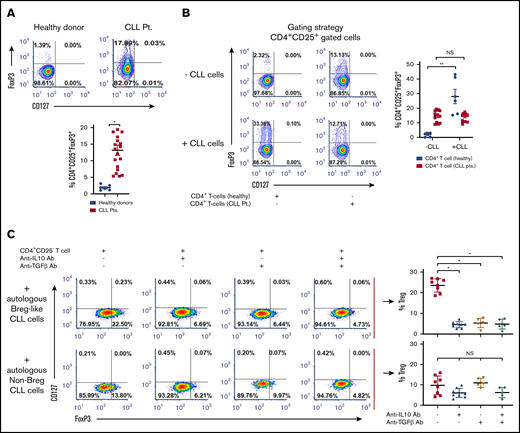
Breg-like CLL cells promote conversion of naive Th cells into Tregs that are mediated by IL-10 and TGF-β. (A) Baseline proportion of CD4+CD25+CD127lo and FoxP3+ Treg cells was compared within PBMCs isolated from healthy donors (n = 6) or patients with CLL (n = 12). (B) CD19+CD5+ cells and autologous naive Th cells (CD4+CD25−) were sorted from patients with CLL (n = 13). CD4+ T cells were also isolated from healthy donors (n = 6). Next, CLL cells were cocultured with either autologous or allogeneic (healthy donor-derived) naive Th cells in a 1:1 ratio that were prestimulated with anti-CD3 and anti-CD28 antibodies. After 3 days of incubation, cells were labeled for Treg markers and analyzed by flow cytometry. (C) To identify the mediators of conversion of naive Th cells to iTregs, a transwell coculture assay was conducted. The upper chamber contained either flow-sorted CD19+CD5+CD24+CD38+ (autologous Breg-like CLL cells) or CD19+CD5+CD24+CD38− (autologous non-Breg) CLL cells from 8 patients with CLL, whereas the lower chamber contained autologous flow-sorted naive Th cells prestimulated with anti-CD3 and anti-CD28 antibodies. In addition, IL-10 or TGF-β neutralizing Ab (alone or in combination with one another) were added to the upper chamber and incubated for another 72 hours. The proportion (%) of Tregs was subsequently analyzed by flow cytometry. Contour or density plots are representative, and compiled data are presented as mean ± SEM, with individual data points overlaid. Each experiment was performed at least twice in duplicate. *P < .05; **P < .001.
Breg-like CLL cells promote conversion of naive Th cells into Tregs that are mediated by IL-10 and TGF-β. (A) Baseline proportion of CD4+CD25+CD127lo and FoxP3+ Treg cells was compared within PBMCs isolated from healthy donors (n = 6) or patients with CLL (n = 12). (B) CD19+CD5+ cells and autologous naive Th cells (CD4+CD25−) were sorted from patients with CLL (n = 13). CD4+ T cells were also isolated from healthy donors (n = 6). Next, CLL cells were cocultured with either autologous or allogeneic (healthy donor-derived) naive Th cells in a 1:1 ratio that were prestimulated with anti-CD3 and anti-CD28 antibodies. After 3 days of incubation, cells were labeled for Treg markers and analyzed by flow cytometry. (C) To identify the mediators of conversion of naive Th cells to iTregs, a transwell coculture assay was conducted. The upper chamber contained either flow-sorted CD19+CD5+CD24+CD38+ (autologous Breg-like CLL cells) or CD19+CD5+CD24+CD38− (autologous non-Breg) CLL cells from 8 patients with CLL, whereas the lower chamber contained autologous flow-sorted naive Th cells prestimulated with anti-CD3 and anti-CD28 antibodies. In addition, IL-10 or TGF-β neutralizing Ab (alone or in combination with one another) were added to the upper chamber and incubated for another 72 hours. The proportion (%) of Tregs was subsequently analyzed by flow cytometry. Contour or density plots are representative, and compiled data are presented as mean ± SEM, with individual data points overlaid. Each experiment was performed at least twice in duplicate. *P < .05; **P < .001.
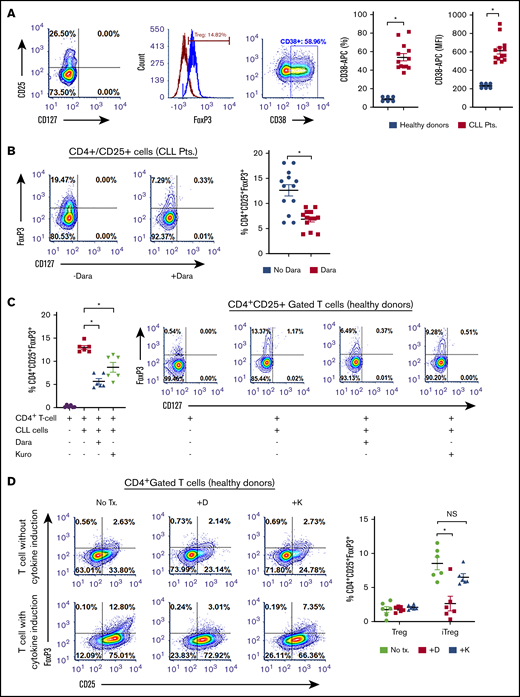
Anti-CD38 agents are lethal to Tregs
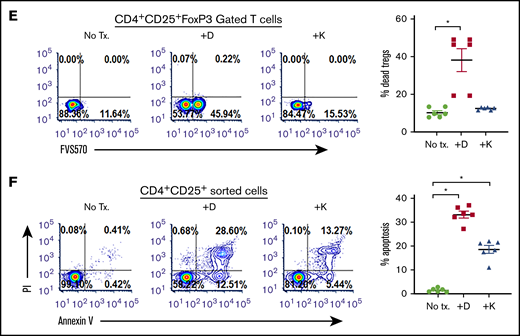
CD38hi Tregs have impressive immunosuppressive capabilities.30,31 We observed a significantly higher proportion of CD38+ Tregs in patients with CLL (54.27% ± 3.91%; MFI, 616.8 ± 36.27) compared with healthy donors (9.07% ± 1.12%; P < .011; MFI, 234.0 ± 10.73; P = .012; Figure 4A). Moreover, a positive correlation between the percentage of CD38+ Tregs and CD38+ B-CLL cells was also observed (r = 0.1; P = .0042; supplemental Figure 2C). As blocking CD38 induces apoptosis in Bregs, we assessed whether Tregs were equally sensitive to CD38-targeting agents. In PBMCs from patients with CLL, ex vivo treatment with daratumumab reduced Tregs by approximately 49% (P = .0286; Figure 4B). To determine whether CD38-targeting agents decreased the formation of iTregs, cocultures containing CLL cells and naive T helper cells from healthy donors were treated with daratumumab or kuromanin. A significant reduction in iTreg formation after exposure to daratumumab (∼50% decrease) or kuromanin (30% decrease; P = .022, Figure 4C) was observed. We also tested whether anti-CD38 agents reduced iTreg formation independent of CLL cells by incubating naive T helper cells from healthy donors with a cytokine cocktail that induces Treg formation. Although daratumumab significantly decreased cytokine-induced iTregs (P = .0115), kuromanin did not (Figure 4D). We questioned whether iTreg depletion was a result of a direct cytotoxic effect of anti-CD38 agents and observed that ex vivo exposure of PBMCs from patients with CLL to daratumumab induced cell death in 38.36% ± 6.06% of FoxP3-gated Tregs (P = .011; Figure 4E). Treg cell death was also confirmed in flow-sorted CD4+CD25+ T cells from PBMCs of patients with CLL, in which daratumumab or kuromanin induced apoptosis in 33.14% ± 1.73% (P = .0009) or 18.04% ± 1.94% (P = .0041) of cells, respectively (Figure 4F). Collectively viewed, our results demonstrate that Tregs from patients with CLL have high CD38 expression and are depleted from the PBMCs of patients with CLL on exposure to daratumumab.
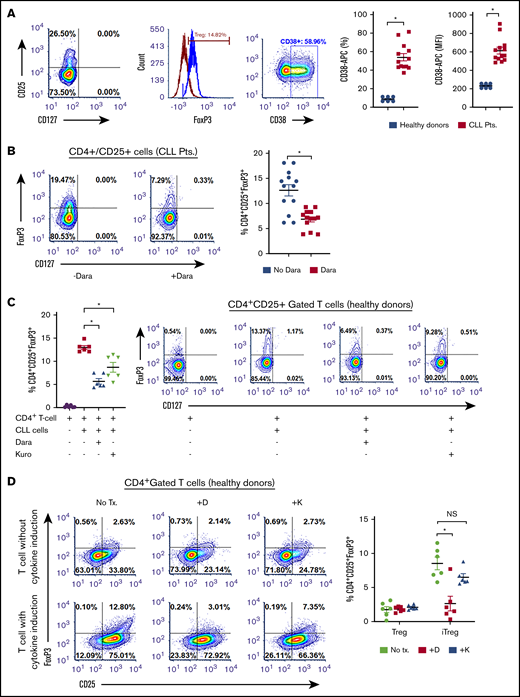
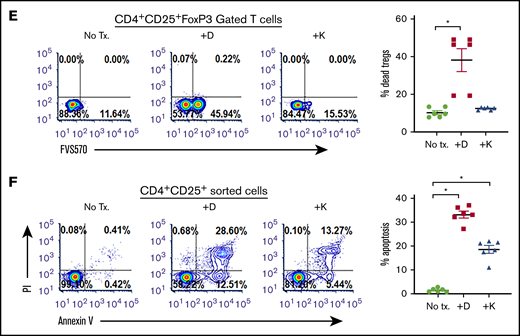
CD38-targeting agents deplete Tregs (A) CD38 expression was measured as percentage expression and MFI on CD4+CD25+CD127lo gated Tregs in PBMCs isolated from healthy donors (n = 6) or patients with CLL (n = 12). Gating strategy is shown, and compiled data of percentage CD38+ cells are presented as mean ± SEM. (B) Using a similar gating strategy, percentage of Tregs was measured ± treatment with daratumumab (1 µg/mL; 72 hours) in PBMCs from 13 patients with CLL. (C) The effect of daratumumab (Dara) or kuromanin (Kuro) on CLL cell-mediated conversion of CD4+CD25− T cells to iTregs was tested. CD19+CD5+-sorted CLL cells were cocultured with CD4+ T cells from healthy donors with or without daratumumab (1 µg/mL) or kuromanin (30 µM) for 72 hours, followed by measurement of iTregs. (D) In a similar manner, the effect of daratumumab (D) or kuromanin (K) was assessed on cytokine induced iTregs (without any CLL cells). (E) The direct cytotoxic effects of daratumumab (D, 1 µg/mL, 24 hours) or kuromanin (K, 30 µM, 24 hours) were measured on Tregs from patients with CLL (n = 6 patients), using a fixable live dead assay. (F) Separately, CD4+CD25+CD127lo sorted cells from 6 patients with CLL were treated with anti-CD38 agents (same concentrations and time as in panel H) and dual-stained with annexin V/PI followed by flow cytometry analysis. Contour plots are representative, and compiled data are presented as mean ± SEM with individual data points overlaid. Each experiment was performed at least twice in duplicate. *P < .05.
CD38-targeting agents deplete Tregs (A) CD38 expression was measured as percentage expression and MFI on CD4+CD25+CD127lo gated Tregs in PBMCs isolated from healthy donors (n = 6) or patients with CLL (n = 12). Gating strategy is shown, and compiled data of percentage CD38+ cells are presented as mean ± SEM. (B) Using a similar gating strategy, percentage of Tregs was measured ± treatment with daratumumab (1 µg/mL; 72 hours) in PBMCs from 13 patients with CLL. (C) The effect of daratumumab (Dara) or kuromanin (Kuro) on CLL cell-mediated conversion of CD4+CD25− T cells to iTregs was tested. CD19+CD5+-sorted CLL cells were cocultured with CD4+ T cells from healthy donors with or without daratumumab (1 µg/mL) or kuromanin (30 µM) for 72 hours, followed by measurement of iTregs. (D) In a similar manner, the effect of daratumumab (D) or kuromanin (K) was assessed on cytokine induced iTregs (without any CLL cells). (E) The direct cytotoxic effects of daratumumab (D, 1 µg/mL, 24 hours) or kuromanin (K, 30 µM, 24 hours) were measured on Tregs from patients with CLL (n = 6 patients), using a fixable live dead assay. (F) Separately, CD4+CD25+CD127lo sorted cells from 6 patients with CLL were treated with anti-CD38 agents (same concentrations and time as in panel H) and dual-stained with annexin V/PI followed by flow cytometry analysis. Contour plots are representative, and compiled data are presented as mean ± SEM with individual data points overlaid. Each experiment was performed at least twice in duplicate. *P < .05.
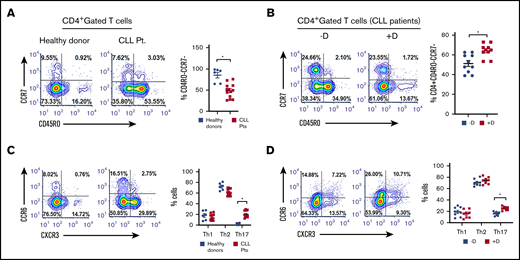
Daratumumab increases the proportion of Th17 cells and promotes expansion of activated cTLs that shows patient tumor-specific cytolytic activity
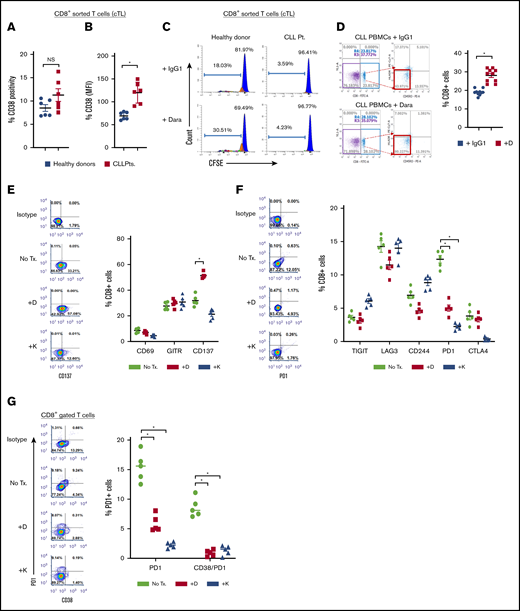
As anti-CD38 agents depleted Tregs and Breg-like CLL cells, we deduced that changes in the proportion of other T-cell subsets may also be evident (workflow/methodology shown in supplemental Figure 4). Assessment of basal T effector levels were found to be lower in PBMCs from patients with CLL (50.05% ± 5.41%) vs healthy donors (86.94% ± 11.19%; P = .014; Figure 5A). After treatment of PBMCs from patients with CLL with daratumumab, a significant increase was noted in the proportion of T effector cells (64.53% ± 2.12%; P = .012; Figure 5B). Notably, significant T effector proliferation was also seen in CD4+ T cells isolated from the PBMCs of patients with CLL and treated ex vivo with daratumumab, but not in isolated CD4+ T cells that were depleted of Tregs (supplemental Figure 5). We also profiled Th1, Th2, and Th17 subsets and found the percentage of Th17 cells to be 11-fold higher in patients with CLL vs healthy donors (P = .004). On treatment with daratumumab, the proportion of Th17 cells from patients with CLL further increased (P = .015), with no changes in Th1 and Th2 subsets seen (Figure 5C-D). In PBMCs depleted of CLL cells and treated with daratumumab, the proportion of Th17 cells remained unchanged, suggesting that daratumumab did not directly increase Th17 cells, but perhaps indirectly through acting on (and killing) CLL cells in the PBMC mixture (supplemental Figure 6). Overall, exposure to daratumumab also increased the proportion of CD4+IL17+ and CD4+IFN-γ+ T cells (supplemental Figure 7). As such, we reasoned that depletion of Tregs and tandem increases in the proportion of Th17 and CD4+IL17+ cells by daratumumab may lead to enhanced cTL antitumor function.32-35 CD38 expression on sorted cTLs from patients with CLL did not differ drastically from that of healthy donors (11.29% ± 1.36% vs 8.27% ± 0.56%; Figure 6A-B), and thus we tested what direct effect daratumumab would have on these cells. Intriguingly, sorted and carboxyfluorescein succinimidyl ester-labeled cTLs from healthy donors treated with daratumumab ex vivo showed a twofold increase in proliferation (P = .026) that was not observed in cTLs isolated from patients with CLL (Figure 6C). Despite lack of proliferation, it remained plausible that cTLs may be activated as a result of daratumumab-mediated reduction in the proportion of Bregs and Tregs, decreased IL-10 and TGF-β, and increased extracellular IFN-γ. Thus, we treated PBMCs from patients with CLL with daratumumab, and noted a modest but significant increase in the proportion of cTLs in the PBMC mixture exposed to daratumumab (28% cTLs) vs control Ab (17% cTLs; P = .03; Figure 6D). In line with this observation, a significant increase in CD137+ cTLs (P = .022; Figure 6E) and decrease in PD-1+ cTL subsets (P = .011; Figure 6F) was also evident in PBMCs from patients with CLL exposed to anti-CD38 agents, indicating an activated cTL phenotype. It has recently been shown that PD1+CD38hiCD8+ T cells are immunosuppressive and mediate resistance to immune checkpoint therapy.36 Assessment of PD1+CD38hi cTLs in PBMCs from patients with CLL treated with daratumumab or kuromanin ex vivo showed a significant decrease in this cTL subtype (P = .001 relative to control; Figure 6G). We also noted that daratumumab exposure resulted in an increased proportion of IL17+ and IFN-γ+ cTLs (supplemental Figure 8), which prompted us to test the hypothesis that these activated/daratumumab-primed cTLs would be highly effective in lysing CLL cells (schema of experiment in Figure 6H). As expected, daratumumab-primed cTLs had greater cytolytic activity toward autologous sorted CLL cells as compared with cTLs that were exposed to an immunoglobin G1 (IgG1) control Ab (P = .019; Figure 6I, compare left side blue dots with left side red squares). To further understand whether these effects were patient-tumor–specific, we exposed daratumumab-primed or IgG1 Ab-exposed cTLs from patients with CLL to either autologous or allogeneic (different patient) CLL cells. Although significant cytolysis was observed in the autologous cocultures that contained daratumumab-primed cTLs vs the IgG-exposed cTLs (P = .026; Figure 6J, compare left side blue dots with left side red squares), nearly equivalent specific lysis between daratumumab-primed cTLs or the IgG-exposed cTLs was observed in the allogeneic coculture (supplemental Figure 6J, compare right side black dots with right side red dots). Altogether, these results indicate to us that daratumumab, through an undefined mechanism, influences the activation state of cTLs and enhances their cytolytic activity toward CLL cells (in a patient tumor-specific manner).
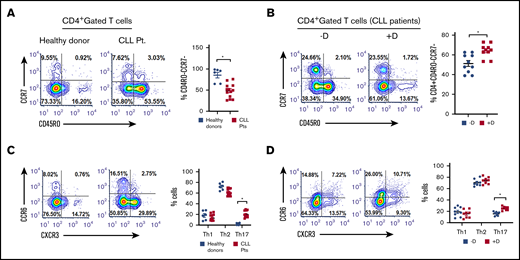
Daratumumab increases the proportion of effector T helper cells and Th17 cells. (A) Naive, effector, central memory, and effector memory Th cell levels measured the PBMCs of healthy donors (n = 6) or patients with CLL (n = 13). These cells were categorized as such based on the following immunophenotypic markers: CD3+CD8-CD4+CCR7+CD45RO− (naive), CD3+CD8−CD4+CCR7−CD45RO− (effector), CD3+CD8−CD4+CCR7+CD45RO+ (CM; central memory)− CD3+CD8−CD4+CCR7−CD45RO+ (EM; effector memory), CD3+CD8−CD4+CCR6−CXCR3+ (Th1), CD3+CD8−CD4+CCR6−CXCR3− (Th2), CD3+CD8−CD4+CCR6+CXCR3− (Th17). (B) The effect of daratumumab (D, 1 µg/mL, 72 hours) in modulating proportion (%) of effector Th cells from patients with CLL (n = 10) was examined. (C) Different T helper cell subset (Th1; Th2 and Th17) were measured in the same healthy donors or patients with CLL, as in panel C. (D) The effect of daratumumab on altering the percentage of Th1, Th2, and Th17 cells from patients with CLL (n = 8) was tested as described in panel B. Contour plots are representative, and compiled data are presented as mean ± SEM with individual data points overlaid. Each experiment was performed at least twice in duplicate. *P < .05.
Daratumumab increases the proportion of effector T helper cells and Th17 cells. (A) Naive, effector, central memory, and effector memory Th cell levels measured the PBMCs of healthy donors (n = 6) or patients with CLL (n = 13). These cells were categorized as such based on the following immunophenotypic markers: CD3+CD8-CD4+CCR7+CD45RO− (naive), CD3+CD8−CD4+CCR7−CD45RO− (effector), CD3+CD8−CD4+CCR7+CD45RO+ (CM; central memory)− CD3+CD8−CD4+CCR7−CD45RO+ (EM; effector memory), CD3+CD8−CD4+CCR6−CXCR3+ (Th1), CD3+CD8−CD4+CCR6−CXCR3− (Th2), CD3+CD8−CD4+CCR6+CXCR3− (Th17). (B) The effect of daratumumab (D, 1 µg/mL, 72 hours) in modulating proportion (%) of effector Th cells from patients with CLL (n = 10) was examined. (C) Different T helper cell subset (Th1; Th2 and Th17) were measured in the same healthy donors or patients with CLL, as in panel C. (D) The effect of daratumumab on altering the percentage of Th1, Th2, and Th17 cells from patients with CLL (n = 8) was tested as described in panel B. Contour plots are representative, and compiled data are presented as mean ± SEM with individual data points overlaid. Each experiment was performed at least twice in duplicate. *P < .05.
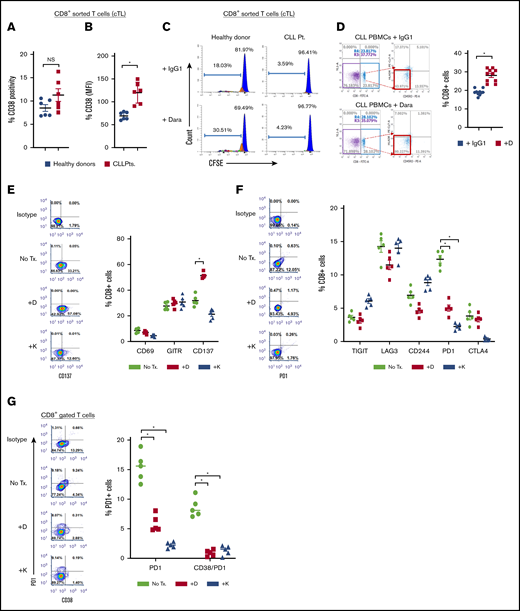
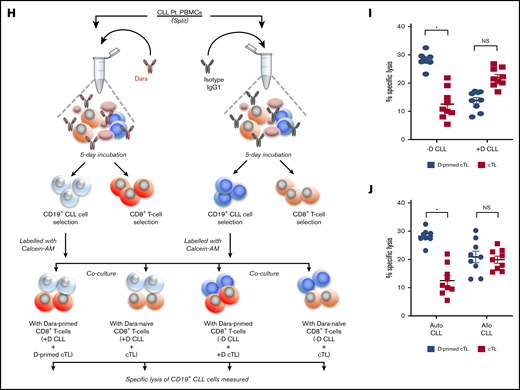
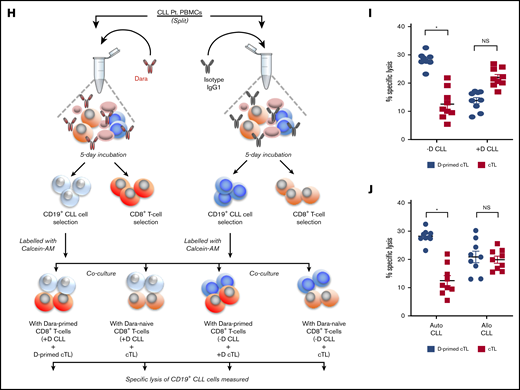
Daratumumab induces expansion of activated and patient-tumor specific cTLs. (A-B) cTLs were sorted from PBMCs of healthy donors (n = 6) and patients with CLL (n = 6), with CD38 levels determined as percentage expression (A) and MFI (B). (C) Flow-sorted cTLs were labeled with carboxyfluorescein succinimidyl ester followed by treatment with daratumumab for 72 hours. cTL proliferation was measured and data were represented as multicolor cell proliferation histograms. (D) PBMCs from patients with CLL were incubated with IgG1 control Ab or daratumumab (1 µg/mL) for 72 hours and CD3+CD8+ T cells were gated to determine percentage of cTLs. (E) Cell surface markers of T-cell activation (CD69, GITR, CD137) and (F) exhaustion (TIGIT, Lag3, CD244, PD1, CTLA4) were measured on CD3+CD8+ gated cTLs from PBMCs of patients with CLL, which were ex vivo treated with daratumumab (D, 1 µg/mL) or kuromanin (30 µM) for 72 hours. Contour plots are representative and compiled data are presented as mean ± SEM with individual data points overlaid. (G) The presence of CD38+PD1+ co-expressing cTLs was measured in CD3+CD8+ gated T cells from PBMCs from patients with CLL treated with daratumumab (1 µg/mL, 72 hours) or kuromanin (30 µM, 72 hours). Data are represented as scatter dot plots of percentage PD1+ cells or percentage CD38+PD1+ cells ± SEM. (H) An illustration of the experimental design in testing daratumumab-primed or unprimed cTL cytolytic response to either untreated CLL cells (−D) or daratumumab-treated CLL cells (+D) from 9 patients is presented. Notably, 7 of these patients were considered as having CD38− disease. (I) Untreated CLL cells (−D) or daratumumab-treated CD19+ CLL cells (+D) were labeled with Calcein-AM and cocultured with daratumumab-primed patient autologous cTLs or unprimed cTLs (exposed to IgG1-isotype control Ab) for 6 hours. Percentage specific lysis of CLL cells was measured as percentage calcein release in supernatant. Calcein-AM released from Triton X-100 treated cells (%) was used as maximum lysis and that from untreated CLL cells was used as spontaneous lysis. (J) Similarly, patient-specific tumor specificity of daratumumab-primed cTLs was measured using Calcein-AM labeled autologous/allogeneic CLL cells. Data are represented as scatter dot plots of percentage mean specific lysis of tumor cells ± SEM. Each experiment was performed at least twice in duplicate. *P < .05.
Daratumumab induces expansion of activated and patient-tumor specific cTLs. (A-B) cTLs were sorted from PBMCs of healthy donors (n = 6) and patients with CLL (n = 6), with CD38 levels determined as percentage expression (A) and MFI (B). (C) Flow-sorted cTLs were labeled with carboxyfluorescein succinimidyl ester followed by treatment with daratumumab for 72 hours. cTL proliferation was measured and data were represented as multicolor cell proliferation histograms. (D) PBMCs from patients with CLL were incubated with IgG1 control Ab or daratumumab (1 µg/mL) for 72 hours and CD3+CD8+ T cells were gated to determine percentage of cTLs. (E) Cell surface markers of T-cell activation (CD69, GITR, CD137) and (F) exhaustion (TIGIT, Lag3, CD244, PD1, CTLA4) were measured on CD3+CD8+ gated cTLs from PBMCs of patients with CLL, which were ex vivo treated with daratumumab (D, 1 µg/mL) or kuromanin (30 µM) for 72 hours. Contour plots are representative and compiled data are presented as mean ± SEM with individual data points overlaid. (G) The presence of CD38+PD1+ co-expressing cTLs was measured in CD3+CD8+ gated T cells from PBMCs from patients with CLL treated with daratumumab (1 µg/mL, 72 hours) or kuromanin (30 µM, 72 hours). Data are represented as scatter dot plots of percentage PD1+ cells or percentage CD38+PD1+ cells ± SEM. (H) An illustration of the experimental design in testing daratumumab-primed or unprimed cTL cytolytic response to either untreated CLL cells (−D) or daratumumab-treated CLL cells (+D) from 9 patients is presented. Notably, 7 of these patients were considered as having CD38− disease. (I) Untreated CLL cells (−D) or daratumumab-treated CD19+ CLL cells (+D) were labeled with Calcein-AM and cocultured with daratumumab-primed patient autologous cTLs or unprimed cTLs (exposed to IgG1-isotype control Ab) for 6 hours. Percentage specific lysis of CLL cells was measured as percentage calcein release in supernatant. Calcein-AM released from Triton X-100 treated cells (%) was used as maximum lysis and that from untreated CLL cells was used as spontaneous lysis. (J) Similarly, patient-specific tumor specificity of daratumumab-primed cTLs was measured using Calcein-AM labeled autologous/allogeneic CLL cells. Data are represented as scatter dot plots of percentage mean specific lysis of tumor cells ± SEM. Each experiment was performed at least twice in duplicate. *P < .05.
Anti-CD38–targeted therapy alters Breg, Treg, and cTL levels in vivo in a CLL-PDX model
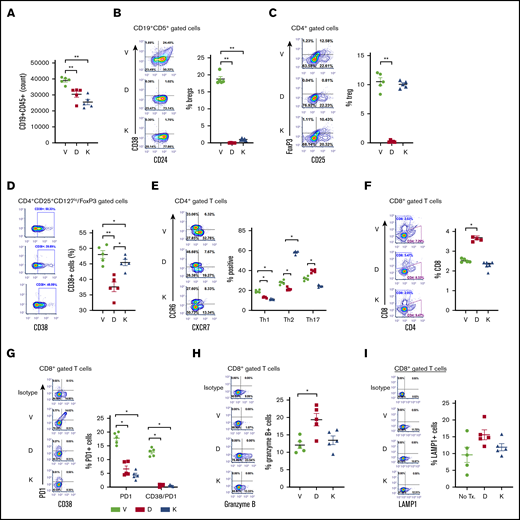
A CLL-PDX mouse model22 was established to determine the effect of daratumumab or kuromanin on B- and T-cell subsets. Compared with mice that received only vehicle, a significant reduction in the absolute number of CD19+CD5+ CLL cells was noted in mice treated with daratumumab or kuromanin (Figure 7A). Notably, Breg-like CLL cells in both daratumumab-treated (P = .00078) and kuromanin-treated mice (P = .00045) were reduced (Figure 7B). Contrastingly, total Tregs were decreased in daratumumab-treated mice (P = .0008), but not in mice treated with kuromanin (Figure 7C). CD38+ Tregs were significantly lower in mice treated with either CD38-targeting agent, but more remarkably so in daratumumab-treated mice (Figure 7D). Examination of T-helper subsets showed that both daratumumab and kuromanin decreased Th1 populations in vivo (Figure 7E); however, changes in Th2 and Th17 subsets were more variable. The proportional change in the percentage of Tregs, Th1, Th2, and Th17 cells and cTLs in spleens isolated from mice treated with CD38-targeted agents was mostly mirrored in the absolute cell count/spleen for these cell types (supplemental Table 1). Although minimal in change, a statistically significant (P = .022) increase in cTLs was noted in mice treated with daratumumab, but not kuromanin (Figure 7F). Notably, PD1+ cTLs and PD1+CD38hi cTLs were significantly lower in mice treated with either CD38-targeting agent compared with vehicle-treated mice (Figure 7G), but it was only in the daratumumab-treated cohort that we also observed an increase in Granzyme B+ or LAMP1+ cTLs, suggesting increased cytolytic capacity of these cells (P = .03 or P = .012, respectively, vs vehicle; Figure 7H-I).
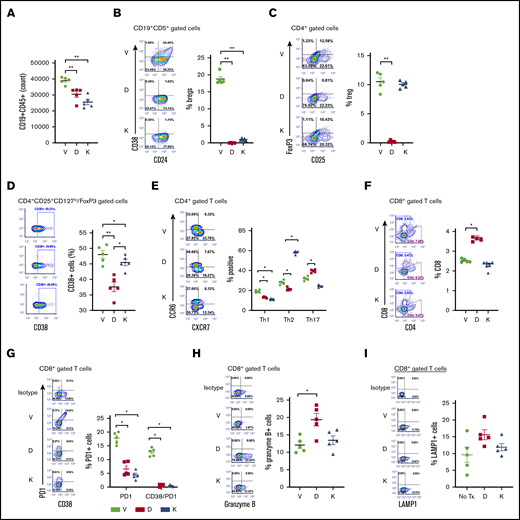
Effects of anti-CD38 agents on Breg-like CLL cells, Tregs, T helper, and cTLs in a CLL PDX model. A short-term PDX-CLL clinical trial concept mouse model was developed via injecting (IV) PBMCs isolated from a patient with CLL (clinical characteristics in Table 1, Pt. 22) into NSG mice. After 24 hours, mice were treated with vehicle (V), daratumumab (D, 20 µg/mL loading dose followed by 10 µg/mL), or kuromanin (K, 20 µg/mL) by IV tail vein injection on postimplantation days 2, 5, and 8. On day 9, mice were sacrificed, with the spleen and blood harvested, followed by human immune cell analysis carried out showing the absolute number of CD19+CD5+ CLL cells (A); Breg-like CLL cells (CD19+CD5+CD24+CD38+) (B); Tregs (CD4+CD25+FoxP3+) cells (C); CD38+Tregs (CD4+CD25+CD127lo/FoxP3 gated cells) (D); T-helper cells Th1, Th2, and Th17 subsets (E); CD8+ cTLs (F); PD1+CD38hiCD8+ cTLs (G); Granzyme B+CD8+ cTLs (H); and LAMP1+CD8+ cTLs (I) were probed and gated for using human antibodies in mouse splenocytes cells. Absolute cell counts for Bregs, Tregs, Th1, Th2, Th17, and cTLs were also determined and are shown in supplemental Table 2. Contour plots are representative and compiled data are presented as mean ± SEM with individual data points overlaid. Each experiment was performed at least twice in duplicate. *P < .05; **P < .001.
Effects of anti-CD38 agents on Breg-like CLL cells, Tregs, T helper, and cTLs in a CLL PDX model. A short-term PDX-CLL clinical trial concept mouse model was developed via injecting (IV) PBMCs isolated from a patient with CLL (clinical characteristics in Table 1, Pt. 22) into NSG mice. After 24 hours, mice were treated with vehicle (V), daratumumab (D, 20 µg/mL loading dose followed by 10 µg/mL), or kuromanin (K, 20 µg/mL) by IV tail vein injection on postimplantation days 2, 5, and 8. On day 9, mice were sacrificed, with the spleen and blood harvested, followed by human immune cell analysis carried out showing the absolute number of CD19+CD5+ CLL cells (A); Breg-like CLL cells (CD19+CD5+CD24+CD38+) (B); Tregs (CD4+CD25+FoxP3+) cells (C); CD38+Tregs (CD4+CD25+CD127lo/FoxP3 gated cells) (D); T-helper cells Th1, Th2, and Th17 subsets (E); CD8+ cTLs (F); PD1+CD38hiCD8+ cTLs (G); Granzyme B+CD8+ cTLs (H); and LAMP1+CD8+ cTLs (I) were probed and gated for using human antibodies in mouse splenocytes cells. Absolute cell counts for Bregs, Tregs, Th1, Th2, Th17, and cTLs were also determined and are shown in supplemental Table 2. Contour plots are representative and compiled data are presented as mean ± SEM with individual data points overlaid. Each experiment was performed at least twice in duplicate. *P < .05; **P < .001.
Discussion
Physiologically, CD38 plays a central role in various immune functions, such as T-cell activation, neutrophil chemotaxis, dendritic cell migration, and monocyte chemokine production.37,38 In patients with CLL, these immune processes are highly dysregulated and underlie the immense degree of overall immune failure observed clinically.1,39-42 In the current study, we investigated the immune cytokine milieu as well as B- and T-cell subsets in patients with CLL, within the context of targeting CD38. Subset analysis in CD38+ vs CD38− patients with CLL revealed not only higher levels of IL-10 and TGF-β but also IFN-γ and IL-2 in plasma from CD38+ patients. IFN-γ+ B cells have been described to promote the innate immune response against intracellular infections by activating dendritic cells via IFN-γ secretion at levels nearly similar to those produced by NK cells.43 Although the significance of IFN-γ+ CLL cells and their increased levels postexposure to anti-CD38 agents is unclear, it is possible that these cells are able to also activate various T-cell subsets (including cTLs) via enhanced secretion of IFN-γ and IL-2.
Studies into B cells with regulatory properties demonstrate that a subset of CLL cells shares remarkable similarity with B10 Bregs.12 We observed that the CD19+CD5+CD24+CD38hi subset of Breg-like CLL cells has a distinct IL-10 secretion pattern compared with CD38lo/non-Breg CLL cells. Indeed, anti-CD38 agents induced apoptosis in Breg-like CLL cells, which was associated with a decrease in IL-10 secretion. Mechanistically, IL-10 and TGF-β secreted by CD38hi CLL-Bregs promotes the expansion/outgrowth of Tregs, more so in comparison with CD38lo CLL cells, which we demonstrated via co-culturing CD38hi or CD38lo CLL cells with patient-autologous naive T cells, and used neutralizing antibodies for IL-10/TGF-β. Jak et al have reported a CD27-CD70-dependent mechanism for CLL-mediated iTreg expansion44 ; however, our observation that iTreg formation is dependent on IL-10/TGF-β is novel (Figure 3C). In line with this, we observed that daratumumab (and to a lesser extent kuromanin) significantly reduced iTreg levels. We questioned whether a reduction in Tregs was due to the direct effect of CD38-targeting agents or because CD38-targeting agents deplete Bregs, reduce IL-10/TGF-B levels, which in turn then decrease Tregs. During this investigation, we made a novel observation that nearly 50% of Tregs from patients with CLL have high CD38 expression compared with Tregs from healthy donors. Furthermore, a significant correlation between the percentage of Tregs and percentage of CD38+ CLL cells present in the PBMCs of patients with CLL was also noted (r = 0.5; P = .012; supplemental Figure 2B). Krejcik et al30 first reported that treatment of patients with multiple myeloma with daratumumab rapidly depletes CD38+ Tregs, and subsequent studies by Feng et al45 showed that blocking CD38 with isatuximab induces Treg apoptosis. Our observations mirror these findings and show that exposure of PBMCs from patients with CLL to daratumumab, ex vivo, induces death of Tregs, a substantial number of which express high levels of CD38.
Among other T-cell subsets, we noted low T effector cell levels in PBMCs from patients with CLL, which increased significantly after exposure to daratumumab. We surmised that an increase in these cells was a result of daratumumab-mediated death of Breg-like CLL cells and Tregs (with concomitant changes in cytokine levels), which act as a stimulus for effector T-cell proliferation,46 and was not a direct stimulatory effect of the drugs itself on T effector cells. Indeed, in PBMCs from patients with CLL, where CD4+ T cells were isolated and Treg depletion was performed, CD38-targeting agents did not increase T effector cell proliferation. In contrast, when all CD4+ T cells (which included Tregs) isolated from PBMCs from patients with CLL were treated with daratumumab or kuromanin, a significant increase in T effector cell proliferation was observed (supplemental Figure 5). Dissection of the various T helper subsets revealed increased Th17 cells in patients with CLL compared with healthy donors, which has previously been reported.1 In CLL, Th17 cells have been suggested to play an antitumor role and are garnering interest in cancer immunotherapy as a whole.47 Chatterjee et al have described the similarities between artificially generated hybrid Th1/Th17 cells and CD4+ T cells from CD38 knockout mice. These 2 T-cell populations display superior antitumor capabilities compared with standard Th1 and Th17 cells or CD4+ T cells from CD38 wild-type mice.47 We noted that daratumumab treatment of CLL PBMCs increased the proportion of Th17 cells; however, the functional aspect of this remain to be defined. Given the changes that were induced by exposure to daratumumab in Tregs and Th17 cells, we also examined cTLs. The functional capacity of cTLs in patients with CLL is severely impaired as a result of defects in immune synapse formation and dysregulated expression of activation or immune checkpoint molecules.1,48 Direct exposure of sorted CLL patient-derived cTLs treated ex vivo with daratumumab did not result in their proliferation, but rather death, in ∼11% (P = .007; supplemental Figure 9), an effect most likely manifested in CD38+ or CD38+PD1+ cTLs (Figure 6G). However, in the context of a PBMC mixture, exposure to daratumumab resulted in the relative expansion of activated cTLs, with increased cytolytic (and patient-tumor-specific) activity.
Alterations of B- and T-cell subsets in patients with multiple myeloma receiving treatment with daratumumab have been described, and our ex vivo as well as in vivo observations in a short-term CLL-PDX model echo these observations.30 As our goal was to study the effect of anti-CD38 treatment on T-cell subsets, establishment of a long-term PDX model was not optimal because of the possibility of xenograft vs host-like disease known to occur in mice injected with CD4+-naive T cells.49,50 Results from our in vivo PDX model were similar to those from our ex vivo CLL patient PBMC analyses, in which daratumumab depleted Breg-like CLL cells and Tregs along with increasing Th17 cells and cTLs with enhanced cytolytic capacity (increased Granzyme B+ or LAMP1+ cTLs).
It is noteworthy that in the majority of experiments, we used kuromanin as a comparator to daratumumab, which inhibits CD38 enzymatic function. The effects of kuromanin were more variable compared with daratumumab in both in vitro/ex vivo as well as in vivo assays. In PBMCs from both CD38+ and CD38− patients with CLL, we observed that kuromanin treatment led to a significant reduction in IL-10+ CLL cells. However, when interrogation of IL-10 was specifically performed in Breg-like CLL cells, kuromanin and even 78c (more potent CD38 enzymatic inhibitor) did not reduce intracellular IL-10 (Figure 2D). This finding warrants further investigation, but suggests that the link between IL-10 expression and CD38 is not linear, and enzymatic blockage of CD38 in Breg-like CLL cells specifically may not be sufficient to downregulate IL-10. Aside from this observation, we noted in our PDX model that kuromanin did not decrease the percentage of Tregs. We surmise that in vivo, kuromanin is a weak CD38 enzymatic inhibitor, and ongoing experiments with 78c will help resolve this.
Although the B-cell compartment is undoubtedly aberrant in patients with CLL, a remarkable degree of dysfunction is also noted in the various T-cell subsets. Our present investigation establishes that therapies aimed at CD38 promote both direct killing of Breg-like CLL cells along with Tregs, with a subsequent enhancement of antitumor T-cell responses. Collectively our observations demonstrate that anti-CD38 agents have an immunomodulatory effect, reprogramming the CLL microenvironment and restoring antitumor immune functionality.
Send data sharing requests via e-mail to the corresponding author.
Acknowledgments
The experiments and analysis carried out in this study were supported in part by the Daniel Foundation of Alabama (A.A.C.-K.), the Predolin Foundation (A.A.C.-K.), and the Mayo Clinic Cancer Center (National Institutes of Health, National Cancer Institute grants CA015083 [A.A.C.-K.] and R01 CA233790-01A1 [A.A.C.-K. and E.C.]).
Authorship
Contribution: A.M., A.A.C.-K., and A.P. provided conception and design; A.M., T.K., L.J.L.-T., N.D., and A.P. developed methodology; A.M., T.K., S. Aulakh, L.J.L.-T., and N.D. acquired data (provided animals acquired and managed patients, provided facilities, etc); A.M., T.K., S. Aulakh, L.J.L.-T., N.D., A.A.C.-K., and A.P. provided analysis and interpretation of data (eg, statistical analysis, biostatistics, computational analysis); A.M., L.J.L.-T., N.D., K.K., E.C., R.M., F.M., J.P.-I., N.L., S. Ailawadhi, T.S., A.A.C.-K., and A.P. provided writing, review, and/or critical scientific revision of the manuscript; A.M., T.K., S. Aulakh, L.J.L.-T., N.D., K.K., A.A.C.-K., and A.P. provided administrative, technical, or material support (ie, reporting or organizing data, constructing databases); and A.A.C.-K. and A.P. provided study supervision.
Conflict-of-interest disclosure: F.M. has received research funding from Tusk Pharmaceuticals and serves as a consultant for Janssen Pharmaceuticals. S. Ailawadhi and A.A.C.-K. receive research funding for clinical trials from Jaansen Pharmaceuticals. E.C. holds a patent for CD38 inhibitors and is a consultant for TeneoBio, Calico, and Cytokinetics. The remaining authors declare no competing financial interests. The current affiliation for S. Aulakh is Department of Internal Medicine and Neuroscience, West Virginia Cancer Institute, Morgantown, WV.
Correspondence: Aneel Paulus, Department of Cancer Biology, Mayo Clinic, 4500 San Pablo Rd, Jacksonville, FL 32224; e-mail: paulus.aneel@mayo.edu; and Asher A. Chanan-Khan, Department of Medicine, Mayo Clinic, 4500 San Pablo Rd, Jacksonville, FL 32224; e-mail: chanan-khan.asher@mayo.edu.