Key Points
PARP-1 is overexpressed in mycosis fungoides, and its high expression correlates with worse overall survival.
Inhibition of PARP-1 in mycosis fungoides induces Blimp-1–mediated p53-dependent apoptosis, highlighting a therapeutic potential.

Abstract
The therapy of advanced mycosis fungoides (MF) presents a therapeutic challenge, and the search for new therapeutic targets is ongoing. Poly(ADP-ribose) polymerase 1 was shown to be upregulated in patients with advanced MF and could be druggable by a new class of chemotherapeutic agents, PARP-1 inhibitors, which are already in clinical trials for other malignancies; however, the role of PARP-1 inhibitors in MF has never been established. We examined the efficacy of talazoparib in the murine model of cutaneous T-cell lymphoma. The cytotoxic effect of talazoparib on Moloney MuLV-induced T-cell lymphoma (MBL2) cells was a result of G2/M cell cycle arrest via the upregulation of p53. The in vivo experiments confirmed the clinical impact of talazoparib on MF tumors. When talazoparib was combined with the histone deacetylase (HDAC) inhibitor, romidepsin, the cytotoxic effect was synergized via downregulation of the DNA-repair genes Fanconianemia complementation group A (FANCA), Fanconi anemia complementation group D2 (FANCD2), and DNA topoisomerase II binding protein 1(TOPBP1)and stimulation of apoptosis via Blimp-1 (PRDM1)/Bax axis. Romidepsin increased the expression of IRF8 and Bcl-6, leading to upregulation of Blimp1and Bax; whereas talazoparib upregulated Blimp-1 and Bax via upregulation of interferon regulatory factor 4 (IRF4), leading to cleavage of caspases 6 and 7. Thus, a combination of talazoparib with romidepsin demonstrated the synergistic antilymphoma effect and warranted further investigation in a clinical trial.
Introduction
Mycosis fungoides (MF) is a clonal malignancy that is derived from cutaneous mature T-lymphocytes. The annual incidence of MF is 10.2 per 1 million persons.1 High relapse rates and a poor prognosis complicate the clinical course and treatment of MF.2 Patients receiving chemotherapeutic agents used for the treatment of advanced stages have an overall response rate of 38% to 45%.3 The median survival of patients with stage IV disease is 18 months, even in the context of multiple lines of therapy.4 Thus, there is a pressing need for new chemotherapeutic agents aimed at decreasing the tumor burden in the advanced stage of MF, and hopefully, extending the lives of patients with MF.
Poly(ADP-ribose) polymerase 1 (PARP-1) is a nuclear enzyme involved in regulating DNA damage and, specifically, single-strand breaks5-7 by making of chains of poly(ADP-ribose) (PAR).5 PAR chains then bind PARP-1 or other nuclear proteins, creating a scaffolding to initiate DNA repair.5,6 If the damage is too severe to make reparation, PARP-1 can also induce apoptosis.5 Baseline expression of PARP-1is usually low, but was shown to be overexpressed in multiple cancers such as leukemias, colorectal carcinoma, and breast carcinoma.5,6 Previously, we have demonstrated the high PARP-1 expression in patients with tumor stage MF and Sezary syndrome.8 Moreover, we found that patients with aggressive disease express PAPR-1 early in the course of their MF, which potentially can be used as a predictive biomarker.8 Initially, PARP inhibitors (PARPi) were indicated only for breast cancer gene (BRCA)–associated tumors such as advanced ovarian cancer and metastatic breast cancer.9 Currently, several PARPi such as iniparib, olaparib, niraparib, veliparib, and talazoparib are in clinical trials for other malignancies such as head and neck squamous cell carcinoma,10,11 gastric cancer,12,13 osteosarcoma,14 and in chronic leukocytic leukemia.15,16 However, remarkably, none of them were tested in cutaneous lymphoma.
Here, we show that talazoparib, a novel selective inhibitor of PARP-1 and PARP-2, kills lymphoma in vitro and in vivo as well as it kills sensitive breast cancer. Moreover, we have demonstrated that the cytotoxic effect of talazoparib can be potentiated when combined with the HDAC inhibitor (HDACi) romidepsin. This combination synergizes the downregulation of the DNA-repair genes and stimulates apoptosis via the Blimp-1 (PRDM1)/Bax axis. We conclude that a combination of talazoparib with romidepsin has a valuable therapeutic potential and should be further explored clinically.
Materials and methods
Cell lines
MBL2 cells, a Moloney MuLV-induced T-cell lymphoma cell line, were confirmed to be of mouse origin, and no mammalian interspecies contamination was detected by IDEXX BioResearch (Columbia, MO). The MBL2 cell line was found to be free of the following pathogens by IDEXX BioResearch: Ectromelia, epizootic diarrhea of infant mice, lymphocytic choriomeningitis virus, lactate dehydrogenase elevating virus, mouse hepatitis virus, murine norovirus, mouse parvoviruys, minute virus of mice, Mycoplasma pulmonis, Mycoplasma sp., Polyoma, pneumonia virus of mice, reovirus type 3, Sendai, and Theiler's murine encephalomyelitis virus. MAB-MD-436 (HTB130) was purchased from ATCC (Manassas, VA). The cell lines were stored in aliquots in liquid nitrogen. Each aliquot was thawed and cultured for no more than 1 week for each experiment. MBL2 were cultured in RPMI-C for 1 week before inoculation into mice.
HUT78, EL4, and MDA-MB-436 were purchased from ATCC. CD4+ MyLa cells were a generous gift from Carlos A. Murga-Zamalloa (University of Illinois Chicago, Chicago, IL).
XTT assay and IC50
Talazoparib was provided by Pfizer Inc. Bexarotene, vorinostat, romidepsin, methotrexte, pralstrexate, and bortezomib were purchased from Sigma-Aldrich (St. Louis, MO). A total of 7 × 104 cells per well in 96-well plates were incubated with a single dose of drugs in 3 replicates. A minimum of 6 doses of each drug were used to establish the half maximal inhibitory concentration (IC50). Cells along with the drug of interest, were coincubated at 37оC for 24 hours. Cells in media without any drugs and media alone were used as internal controls. After 24 hours, 2,3-Bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide inner saltsodium 3'-[1-[(phenylamino)-carbonyl]-3,4-tetrazolium]-bis(4-methoxy-6- nitro)benzene-sulfonic acid (XTT) Cell Viability assay (ThermoFisher Scientific, Waltham, MA) was performed. Briefly, 25 μL of XTT/N-methyl dibenzopyrazine methyl sulfate solution was added to each well containing 100-μL samples and was coincubated for 2 hours at 37оC in a CO2 incubator. Absorbance was measured at 450 nm (Spectra Max 340PC; Molecular Devices). Cellular IC50 values were determined for each drug using a dose-response curve in Prism software (GraphPad Software, Inc., La Jolla, CA).
Determination of the additive and synergistic effect
Synergism and additive effect was evaluated using the median effect equation of Chou and Talalay17 to generate a combination index and isobologram at 50% effect level.
IHC
Formalin-fixed paraffin-embedded skin biopsies from 25 patients with MF were collected from the tissue bank at the University of Pittsburgh. Commercial antibodies were used for PARP-1 staining (sc-8007, dilution 1:400; Santa Cruz Biotechnology, Inc., Dallas, TX). Human placenta and tonsils were used as positive controls for PARP-1. After deparaffinization, EDTA heat-induced epitope retrieval was conducted for 60 minutes at 95°C (Laboratory Vision EDTA buffer, AP-9004-500; ThermoFisher Scientific). Endogenous peroxidase activity was quenched with 3% hydrogen peroxide for 10 minutes. Prior to blocking with Avidin/Biotin Blocking Kit (BS966L, Background Sniper; Biocare Medical, Concord, CA), slides were incubated with normal mouse serum for 20 minutes. Slides were incubated with primary antibodies for 30 minutes, 10 minutes with Mach 3 Mouse AP Probe, 10 minutes with Mach 3 Mouse AP-Polymer (M3M532H; Biocare Medical), and then 4 minutes with Streptavidin-AP Label (Ap605H; Biocare Medical). Warp Red Chromagen was applied for 15 minutes (WR8065; Biocare Medical). Counterstaining was performed with Harris Hematoxylin for 15 seconds.
The stains for CD1a, CD3, CD4, CD8, CD5, CD7, and Ki67 of the same sections were available as a part of routine clinical care. The slides stained for PARP-1 were analyzed in parallel with the aforementioned markers. The CD3+CD1a−CD4+Ki67+ cells negative for CD5 ± CD7 were considered to be malignant18 when CD3+CD1a−CD4+CD5+CD7+ cells were determined to be reactive. Immunostaining of PARP-1 was scored using a scoring system developed by Allred et al.19 The percentage of stained cells across the whole sample was estimated on a scale of 0 to 100. Staining intensity was rated as negative (0), weak (1), intermediate (2), or strong (3). A total immunohistochemistry (IHC) score was calculated by adding the percentage multiplied by the intensity score. The range of values was 0 to 300.
RNA-sequencing analysis and identification of differentially expressed mRNAs
Total RNA was extracted using the TRIzol reagent (ThermoFisher Scientific) method immediately after cells were treated with drugs. DNA contamination was eliminated using DNase. Library construction was performed by TruSeq Stranded mRNA. Randomly fragment-purified RNA were isolated for short read sequencing. Fragmented RNA were reverse transcribed into cDNA. Adapters were ligated onto both ends of the cDNA fragments. After amplifying fragments using polymerase chain reaction, the fragments with sizes between 200 and 400 bp were selected. For pair-end sequencing on Illumina NovaSeq6000 S4 (2 × 150 bp), both ends of the cDNA were sequenced by the read length (20 million read depth; 40 million total reads), which were performed by Psomagen Inc. (Rockville, MD). The quality control of the sequenced raw read were analyzed. Overall quality of the reads, total bases, total reads, guanine-cytosine content (%), and basic statistics were calculated. Artifacts such as low-quality reads, adaptor sequence, contaminant DNA, or polymerase chain reaction duplicates were removed to reduce biases in analysis. Trimmed reads were mapped to a reference genome with HISAT2. Transcripts were assembled by StringTie with aligned reads. A fold change cutoff of log2 ≤1.5 or ≥0.5 and a P value cutoff of P < .05 were adopted to select differentially expressed and significantly regulated gene sets. Functional enrichment by Gene Ontology analysis was performed to infer potential biological processes and pathways of methylation-associated genes through the IPA Bioinformatics Platform (Qiagen, Venlo, The Netherlands). Results with P < .05 were considered as significant functional categories.
Mouse model of cutaneous T-cell lymphoma
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh (protocol #18093600). Mice were maintained under specific pathogen-free conditions according to guidelines issued by the University of Pittsburgh. The MBL2 cutaneous T-cell lymphoma model was reproduced as previously described.20,21 Briefly, 6- to 8-week-old mice were topically treated with 2,4-dinitrofluorobenzene (0.5% in a vehicle consisting of 4:1 (volume-to-volume ratio) acetone and olive oil, 0.2 mL per ear) (Fluka; Sigma-Aldrich) on dorsal ear skin over the injection site within 10 minutes before tumor cell inoculation. Phosphate-buffered saline (PBS)–washed MBL2 cells (4 × 105 in 20 µL of PBS) were injected into the dermal space under the central dorsal surface of the ears and above the cartilage plane using a 32-gauge needle. Tumor growth was assessed as maximum ear thickness using a digital caliper. Mice were treated on day 14 of the tumor implantation. The drugs were administrated daily for 7 days. In the first set of experiments, PBS (control) and talazoparib were administered intraperitoneally. In the second set of experiments, talazoparib was administered orally; whereas romidepsin and PBS were administered via the intraperitoneal route. The mice were euthanized 3 days after the last injection, on day 24 of implantation, to adjust for immune-mediated cytotoxicity.
Flow cytometry
Cell staining was performed with anti-Blimp1 (goat polyclonal to RPDM1/Blimp-1; Abcam, Cambridge, MA). Annexin V-FITC (BD Bioscience, San Jose, CA) and propidium iodide (Sigma-Aldrich) were used at 2 µL per 50 000 cells. After 15 minutes of coincubation at room temperature in the dark, cells were analyzed by flow cytometry immediately in the appropriate amount of PBS. Data were acquired on a LSRII using CellQuest software (BD Bioscience). FlowJo (Tree Star Inc., Ashland, OR) was used for analysis of the flow cytometric data.
Cell cycle analysis
After treatment, MBL2 cells were pulsed with 10 µM EdU for 2 hours prior to detection with Alexa Fluor 647 azide (Click-iT EdU assay; ThermoFisher Scientific). Cells were subsequently stained by adding 2 drops of FxCycle Violet Ready Flow Reagent (ThermoFisher Scientific) and incubated for 30 minutes at 25°C. Data were acquired on an LSRII Flow Cytometer using a 405 laser and 440/50 nm emission filter.
Confocal microscopy with Hoechst 33258
After treatments with talazoparib for 24 h, MBL2 cells were washed with PBS, and stained with Hoechst 33258 fluorochrome (5 μg/mL) for 10 minutes at room temperature in the dark. After washing twice more with PBS, the Hoechst-stained nuclei were imaged with a confocal microscope (Nikon A1 S232; Nikon, Tokyo, Japan).
Western blotting
MBL2 cells were lysed in RIPA Buffer (#R0278; Sigma-Aldrich) in presence of Protease Inhibitor Cocktail (#P8340; Sigma-Aldrich). Proteins levels were determined by using Pierce BCA Protein Assay Kit (#23225; ThermoFisher Scientific). Proteins were separated using 10% Mini-PROTEAN TGX Precast Protein Gel (#4561033; Bio-Rad Laboratories, Hercules, CA), following transferring to polyvinylidene difluoride (PVDF) membranes (#1620177; Bio-Rad Laboratories). Unspecific binding was blocked using LI-COR Odyssey Blocking Buffer (#92740000; BioAgilytix, Durham, NC). The PVDF membranes where then incubated with the rabbit antimouse antibodies and the appropriate secondary antibodies (IRDye 800CW Goat anti-Rabbit IgG, #92532211; BioAgilytix). The following antibodies were used as primary antibodies: BCL2L11-Bim (#2933, 1:500), Caspase-6 (#9762, 1:250), Caspase-7 (#9492T, 1:500), Caspase-9 (#9504T, 1:500), CDC2 (#28439, 1:500), Cyclin D1 (#2978T, 1:500), and G6PD (#8866, 1:500). All primary antibodies were purchased from Cell Signaling (Danvers, MA). To decrease noise, membranes were washed 3 times in PBS–0.1% Tween 20. Images were obtained by using a LI-COR Scanner (BioAgilytix) in 700 and 800 channels. Before the same membrane was reused, it was stripped using Restore PLUS western Blot Stripping Buffer (#46430; ThermoFisher Scientific).
Statistical Analysis
Statistical analyses were based on the calculation of arithmetic mean and standard deviation. The difference between 2 means was compared by a 2-tailed unpaired Student t test without the assumption of equal variances. The difference between more than 2 means was compared by 1-way analysis of variance with Tukey’s posttest. P < .05 was considered statistically significant. Overall survival (OS) was defined as the time from the first day of diagnosis to death from any cause. Patients without an event in OS were censored on the last day with valid information for the respective endpoint. OS was estimated according to Kaplan-Meier and compared by log-rank (Mantle-Cox) trend test. Multivariate analysis was conducted with the use of Cox proportional hazard model to estimate hazard ratios for evolving an event. The nominal significance level was at 0.05 2-sided. SPSS and Prism software was used for statistical analyses.
Results
High PARP-1 expression in MF correlates with poor prognosis
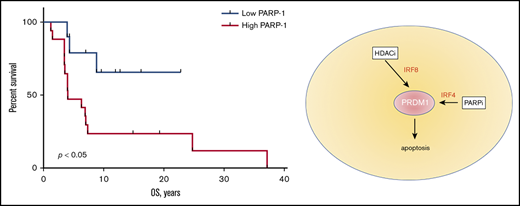
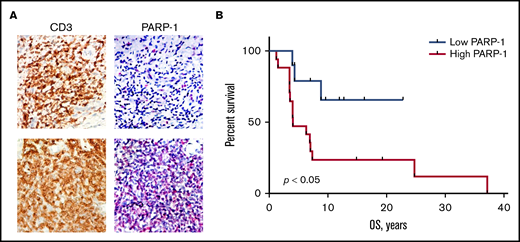
PAPR-1 is overexpressed in many hematologic and solid tumors; however, little is known about PARP-1 in MF. We have previously demonstrated that the patients with aggressive MF express PARP-1 early in the course of their lymphoma, which potentially can be used as a predictive biomarker.8 We have investigated if the expression of PARP-1 on tumor cells (Figure 1A) correlates with the OS of patients with MF. The tissue samples from 9 alive and 16 deceased patients with MF were evaluated for PARP-1 expression: 13 patients with patches, 7 patients with plaques, and 5 patients with tumors. The IHC score varied from 17 to 204. The median IHC score of 50 was chosen as a cutoff. The patients with high PARP-1 IHC score (>50) had a median OS of 4.0 years, whereas the OS was undefined (P < .01) in patients whose PARP-1 IHC score in malignant lymphocytes was low (<50) (Figure 1B). We performed multivariate analysis adjusted for the clinical characteristics to identify contributing factors in relationship to PARP-1 expression. The only statistically significant increase in HR of PARP-1 for shorter OS was observed in elderly patients (HR, 1.083; 95% confidence interval [95% CI], 1.010-1.160; P < .05).
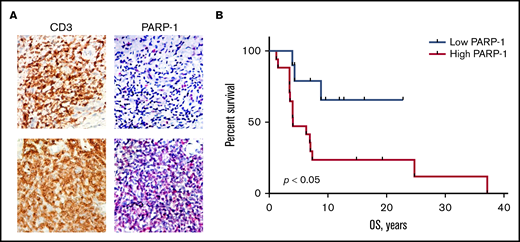
High PARP-1 expression in MF correlates with poor OS. (A) Representative IHC samples of the skin biopsy from a patient with MF with equal CD3 and low PARP-1 (top panels) and high PARP-1 (bottom panels) expression (original magnification, ×40). (B) Kaplan-Meier curves of OS (years) of patients with MF having a low PARP-1 IHC score (<50) vs high PARP-1 IHC score (>50) (n = 25). Log-rank (Mantel-Cox) test (P < .05).
High PARP-1 expression in MF correlates with poor OS. (A) Representative IHC samples of the skin biopsy from a patient with MF with equal CD3 and low PARP-1 (top panels) and high PARP-1 (bottom panels) expression (original magnification, ×40). (B) Kaplan-Meier curves of OS (years) of patients with MF having a low PARP-1 IHC score (<50) vs high PARP-1 IHC score (>50) (n = 25). Log-rank (Mantel-Cox) test (P < .05).
Talazoparib arrests the cell cycle of lymphoma cells at G2/M
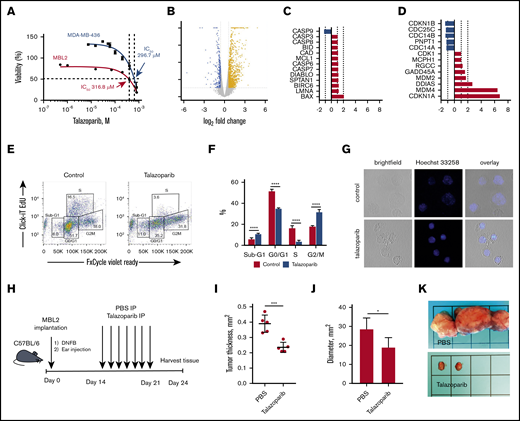
For our in vitro and in vivo experiments, we chose murine CD4+ cell line MBL2 for 2 reasons: MBL2 expressed PARP-1, whereas another popular CD4+ lymphoma cell line, MyLa, did not (supplemental Figure 1), and the MBL2 model is a very well-established orthotopic model closely mimicking tumor stage of MF in immunocompetent animals.20,22 We performed XTT assay to determine the IC50 of talazoparib in MBL2 cells. We found that IC50 for talazoparib was 316.8 μM. We compared those values with data obtained from MDA-MB-436 (BRCA1/2 mutant) cells, which were reported as being a highly sensitive cell line to talazoparib.23 We found that IC50 of talazoparib in MBL2 were comparable with MDA-MB-436, indicating high sensitivity of MBL2 to talazoparib (Figure 2A). Next, we performed RNA sequencing to identify the changes in transcriptome associated with the effect of talazoparib at 25% inhibitory concentration (IC25) on the survival properties of MBL2 cells (Figure 2B). A total of 740 genes were upregulated and 211 genes were downregulated in MBL2 cells (>1.5 times; P < .05). Among the most significant pathway were tRNA charging, protein kinase A signaling, and glycolysis I pathways; all of which were downregulated. Comparison of the transcriptome of apoptosis and cell cycle pathways showed upregulation of BAX in apoptosis and GADD45A, MDM2, DDIAS, MDM4, and CDKN1A in cell cycle pathway (Figure 2C-D). The upregulation of CDKN1a (p21), which plays a critical role in the control of the cell cycle, was more than sixfold. ClickI-EdU assay, coupled with FxCycle Violet, demonstrated that talazoparib arrest the cell cycle at G2/M (Figure 2E-F). The accumulation of a DNA breakage in MBL2 after exposure to talazoparib is demonstrated by Hoechst 33258 fluorescent staining (Figure 2G). The efficacy of talazoparib was confirmed in vivo (Figure 2H-I), where the drug efficiently decreased the size of cutaneous tumors as well as regional lymph nodes involved by lymphoma (Figure 2J-K). Mice did not exhibit any signs of toxicity (such as weight loss, decrease appetite, diarrhea, etc.) while treated with talazoparib.
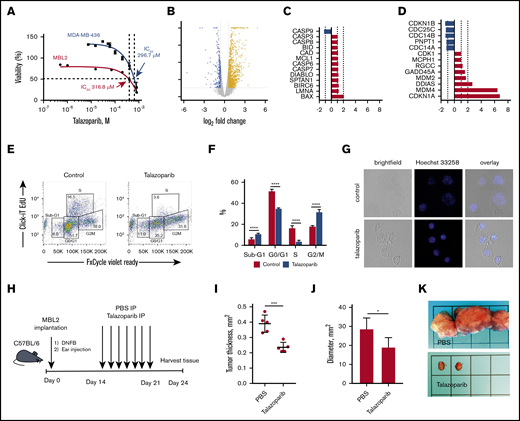
Talazoparib arrests the cell cycle of lymphoma cells at G2/M. (A) Dose-response cell-viability curves at 24 hours for MBL2 and MDA-MB-436. (B) A volcano plot showing changes in transcriptome associated with the effect of talazoparib at IC25 on the survival properties of MBL2 cells identified by RNA sequencing. (C) The transcriptional changes in the apoptosis pathway in MBL2 cells treated with IC25 of talazoparib. (D) The transcriptional changes in the cell cycle pathway in MBL2 cells treated with IC25 of talazoparib. (E) Cell cycle assessment after treatment of MBL2 cells with IC25 of talazoparib. Click-iT EdU with FxCycle Violet Ready Assay. Representative flow cytometry. (F) The differences between the phases of cell cycle of cells treated with IC25 of talazoparib and controls. ****P < .0001. (G) Hoechst 33258 fluorescent staining to show DNA breakage in MBL2 after exposure to talazoparib. Representative images, confocal macroscopy (original magnification, ×100). (H) An outline of the murine experiment. (I) The measurement of the thickness of the orthotopic tumors 21 days after treatment with talazoparib; n = 5 mice per group. ***P < .001 (J) The measurement of the diameter of the regional lymph nodes 21 days after treatment with talazoparib; n = 5 mice per group. *P < .05 (K) Representative images of regional lymph nodes treated with PBS or talazoparib.
Talazoparib arrests the cell cycle of lymphoma cells at G2/M. (A) Dose-response cell-viability curves at 24 hours for MBL2 and MDA-MB-436. (B) A volcano plot showing changes in transcriptome associated with the effect of talazoparib at IC25 on the survival properties of MBL2 cells identified by RNA sequencing. (C) The transcriptional changes in the apoptosis pathway in MBL2 cells treated with IC25 of talazoparib. (D) The transcriptional changes in the cell cycle pathway in MBL2 cells treated with IC25 of talazoparib. (E) Cell cycle assessment after treatment of MBL2 cells with IC25 of talazoparib. Click-iT EdU with FxCycle Violet Ready Assay. Representative flow cytometry. (F) The differences between the phases of cell cycle of cells treated with IC25 of talazoparib and controls. ****P < .0001. (G) Hoechst 33258 fluorescent staining to show DNA breakage in MBL2 after exposure to talazoparib. Representative images, confocal macroscopy (original magnification, ×100). (H) An outline of the murine experiment. (I) The measurement of the thickness of the orthotopic tumors 21 days after treatment with talazoparib; n = 5 mice per group. ***P < .001 (J) The measurement of the diameter of the regional lymph nodes 21 days after treatment with talazoparib; n = 5 mice per group. *P < .05 (K) Representative images of regional lymph nodes treated with PBS or talazoparib.
HDACi, romidepsin, acts synergistically with talazoparib, killing lymphoma cells in vitro
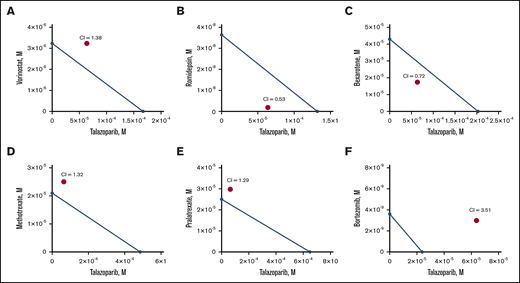
Next, we determined the MBL2 IC50 for the following chemotherapeutic drug currently approved or actively used in clinical practice for the treatment of cutaneous T-cell lymphoma: bexarotene, vorinostat, romidepsin, methotrexate, pralatrexate, and bortezomib (Table 1). We have also estimated IC50 for a human lymphoma cell line, HUT78. Bortezomib demonstrated the lowest IC50, whereas talazoparib had the highest IC50 comparable to methotrexate. Besides pralatrexate and bortezomib, the IC50 values of which were comparable for MBL2 and HUT78, HUT78 cells were more sensitive to talazoparib, bexarotene, vorinostat, romidepsin, and methotrexate than MBL2. The additive interaction was defined as the effect of 2 chemicals, which is equal to the sum of the impact of the 2 chemicals taken separately. The synergistic interaction was determined as the effect of 2 compounds taken together that is greater than the sum of their separate effect at the same doses. Talazoparib at IC25 (63.8 μM) was used for those experiments to determine the synergism of 2 medications (talazoparib plus another agent) (Figure 3A-F). We found that the synergistic effect was observed when talazoparib was combined with romidepsin (Figure 3B) and bexarotene (Figure 3C). However, the combination index was lower in the combination of talazoparib with romidepsin, and that was why that combination was used for further studies.
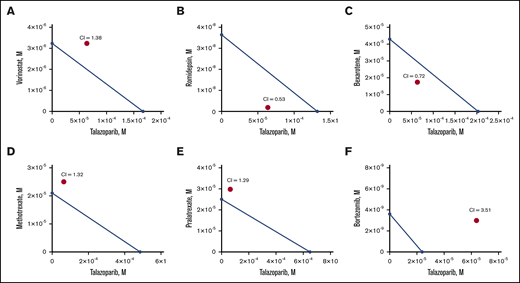
Synergism of combination of talazoparib and romidepsin in vitro. Isoblogram analysis of the effect of fixed molar ration combinations of talazoparib with vorinostat (A), romidepsin (B), bexarotene (C), methotrexate (D), pralatrexate (E), and bortezomib (F) during 24 hours of treatment, assessed by XTT assay. The diagonal line indicates equipotent drug combinations that results in 50% effect level if effects are additive. The red dot represents the real-life result of the drug combination. If the red dot falls beneath the line, it represents synergism; if it falls above the line, it represents antagonism. Combination index (CI) is a numeric measure of synergy. A combination index of 0.30 to 0.70 indicates synergism; 0.70 to 0.84 indicates moderate synergism; 0.85 to 0.90 indicates slight synergism; 0.90 to 1.10 indicates nearly additive effects; and >1.00 indicates antagonism.
Synergism of combination of talazoparib and romidepsin in vitro. Isoblogram analysis of the effect of fixed molar ration combinations of talazoparib with vorinostat (A), romidepsin (B), bexarotene (C), methotrexate (D), pralatrexate (E), and bortezomib (F) during 24 hours of treatment, assessed by XTT assay. The diagonal line indicates equipotent drug combinations that results in 50% effect level if effects are additive. The red dot represents the real-life result of the drug combination. If the red dot falls beneath the line, it represents synergism; if it falls above the line, it represents antagonism. Combination index (CI) is a numeric measure of synergy. A combination index of 0.30 to 0.70 indicates synergism; 0.70 to 0.84 indicates moderate synergism; 0.85 to 0.90 indicates slight synergism; 0.90 to 1.10 indicates nearly additive effects; and >1.00 indicates antagonism.
Proapoptotic synergistic effect of the combination of talazoparib with romidepsin in vitro and in vivo
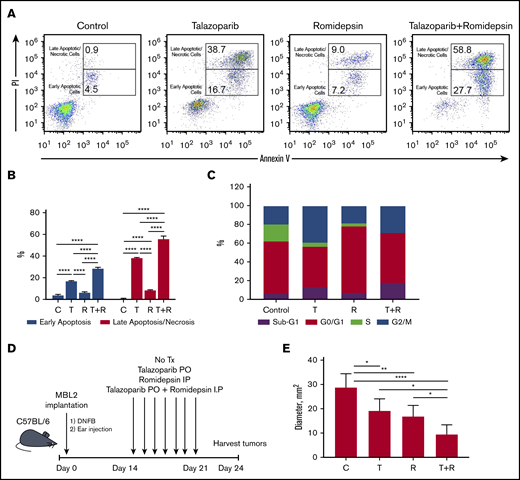
The treatment of MBL2 cells with a combination of talazoparib and romidepsin was associated with increases in early apoptosis (with retention of the cell membrane) as well as increases in late apoptosis/necrosis (with compromised cell membrane) (Figure 4A-B). Interestingly, although the contribution of the combination therapy to early apoptosis seems to be additive, the induction of late apoptosis after combination therapy is an effect of synergism. The analysis of the cell cycle after the treatment with talazoparib and romidepsin demonstrated complete disappearance of the S phase of the cell cycle, an expansion of sub-G1 because of an increase of apoptotic cells, and expansion of G2/M, indicating the arrest of the cell cycle at this phase (Figure 4C). The effect of romidepsin on the cell cycle was different in comparison with talazoparib and is because of the arrest of G0/G1. The best clinical response was found to be in mice that received a combination treatment (an experimental schema presented in Figure 4D) in comparison with mice that received drugs separately (Figure 4E).
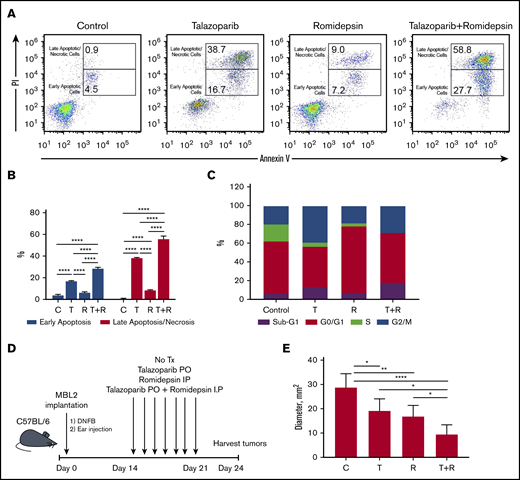
Proapoptotic synergistic effect of combination of talazoparib with romidepsin in vitro and in vivo. (A) Flow cytometry-based analysis of early (Annexin V+PI−) and late (Annexin V+PI+) apoptosis in MBL2 cells treated with talazoparib alone, romidepsin alone, or talazoparib with romidepsin. (B) Analysis of early and later apoptosis in flow cytometry–based assay (Annexin V and PI) in MBL2 cells treated with talazoparib (T) alone, romidepsin (R) alone, or talazoparib with romidepsin (T+R). ***P < .001, ****P < .0001. (C) Analysis of cell cycle arrest with Click-iT EdU with FxCycle Violet Ready Assay. MBL2 cells treated with talazoparib alone (T), romidepsin (R), or a combination of talazoparib with romidepsin (T+R). (D) The outline of the experiment. (E) The effect of combination of talazoparib and romidepsin on the diameter of regional lymph nodes 24 days after treatment. *P < .05, **P < .01, ****P < .0001. C, control.
Proapoptotic synergistic effect of combination of talazoparib with romidepsin in vitro and in vivo. (A) Flow cytometry-based analysis of early (Annexin V+PI−) and late (Annexin V+PI+) apoptosis in MBL2 cells treated with talazoparib alone, romidepsin alone, or talazoparib with romidepsin. (B) Analysis of early and later apoptosis in flow cytometry–based assay (Annexin V and PI) in MBL2 cells treated with talazoparib (T) alone, romidepsin (R) alone, or talazoparib with romidepsin (T+R). ***P < .001, ****P < .0001. (C) Analysis of cell cycle arrest with Click-iT EdU with FxCycle Violet Ready Assay. MBL2 cells treated with talazoparib alone (T), romidepsin (R), or a combination of talazoparib with romidepsin (T+R). (D) The outline of the experiment. (E) The effect of combination of talazoparib and romidepsin on the diameter of regional lymph nodes 24 days after treatment. *P < .05, **P < .01, ****P < .0001. C, control.
The synergistic effect of combination of talazoparib with romidepsin is mediated via Blimp1-Bim axis
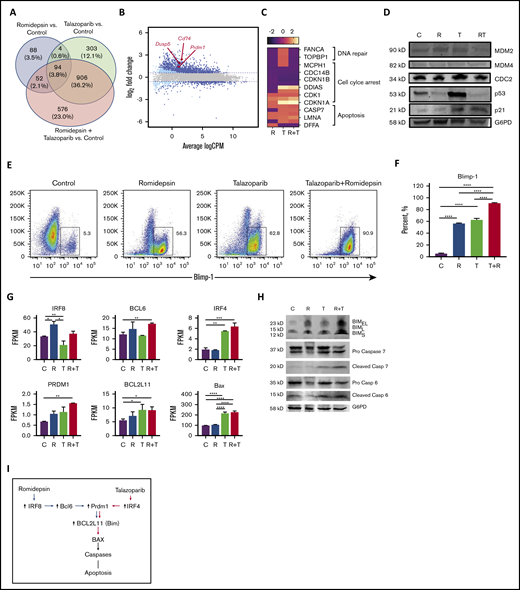
To explore the molecular mechanisms by which the romidepsin potentiate the effect of talazoparib, RNA sequencing analysis of MBL2 cell lines treated with talazoparib, romidepsin, or talazoparib plus romidepsin was performed. Forty percent of the 2023 genes in combination were from talazoparib, whereas only 5.9% of the genes deregulated during the treatment with the combination of talazoparib plus romidepsin were from the effect of romidepsin (Figure 5A). A total of 94 genes were presented in all 3 treatment groups and included upregulation of PRDM1, DUSP5, and CD74, among others (Figure 5B; supplemental Table 1). Analysis of genes responsible for DNA repair, cell cycle arrest, and apoptosis demonstrated the contribution of romidepsin to downregulation of DNA repair, the synergistic effect of romidepsin and talazoparib on downregulation of genes of cell cycle leading to cell cycle arrest, and activation of apoptosis by talazoparib predominantly (Figure 5C). Western blot performed on several survival or antiapoptotic factors such as MDM2, MDM4, CDC2, p53, and p21 showed remarkable upregulation of p53 in the talazoparib group only (Figure 5D). Although no p53 upregulation was found in the combination group, an increased expression of p53 downstream target, p21, indicated the activation of the MDM2/p53 pathway in this group as well. Flow cytometry demonstrated significant upregulation of Blimp-1 (Figure 5E) and its gene PRDM1 (Figure 5F) in the case of the combination of romidepsin with talazoparib. Contribution to this upregulation of romidepsin was mediated via the upregulation of IRF8 and BCL6 (Figure 5G); whereas the contribution of talazoparib was mediated via upregulation of IRF4 (Figure 5G). Both drugs, talazoparib and romidepsin, as well as their combination, led to the accumulation of Bim (Figure 5H). Interestingly, although BCL2L11 was upregulated in talazoparib and the combination group, Bim (the protein transcribed from BCL2L11), was found to be in cells treated with romidepsin. Activation of Bax with subsequent cleavage of caspase 6 and 7 (Figure 5H) contributed to the effect of talazoparib in the combination of talazoparib with romidepsin. A proposed schema of activation of apoptosis is shown in Figure 5I.
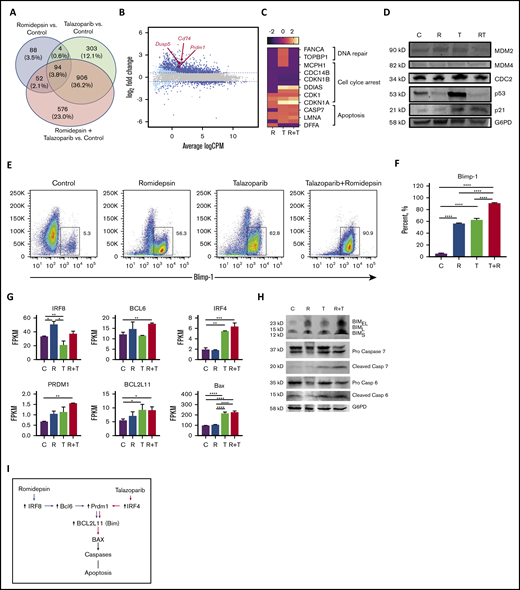
The synergistic effect of the combination of talazoparib with romidepsin is mediated via Blimp1. (A) Venn diagram. (B) Smear plot of transcriptional changes after talazoparib plus romidepsin demonstrates selected genes presented in all 3 groups. (C) The transcriptional changes in DNA repair, cell cycle arrest, and apoptosis pathways in MBL2 cells treated with talazoparib and romidepsin. (D) Western blots for expression of proteins MDM2, MDM4, CDC2, p53, p21, and G6PD from MBL2 cells without treatment (C) or treated with romidepsin (R), talazoparib (T), or romidepsin plus talazoparib (R+T). (E-F) The Blimp-1 expression after treatment. Representative flow (E) and statistical analysis (F). ****P < .001. (G) FPKM of IRF8, BCL6, IRF4, PRDM1, BCL2L11, and BAX after treatment with romidepsin (R), talazoparib (T), and romidepsin plus talazoparib (R+T). MBL2 cells without any treatment were used as controls (C). RNASeq. *P < .05, **P < .01, ***P < .001, ****P < .0001. (H) Corresponding western blots of proteins after treatment with romidepsin (R), talazoparib (T), and romidepsin plus talazoparib (R+T). (I) Proposed schema of activation of apoptosis during the combination therapy with romidepsin and talazoparib.
The synergistic effect of the combination of talazoparib with romidepsin is mediated via Blimp1. (A) Venn diagram. (B) Smear plot of transcriptional changes after talazoparib plus romidepsin demonstrates selected genes presented in all 3 groups. (C) The transcriptional changes in DNA repair, cell cycle arrest, and apoptosis pathways in MBL2 cells treated with talazoparib and romidepsin. (D) Western blots for expression of proteins MDM2, MDM4, CDC2, p53, p21, and G6PD from MBL2 cells without treatment (C) or treated with romidepsin (R), talazoparib (T), or romidepsin plus talazoparib (R+T). (E-F) The Blimp-1 expression after treatment. Representative flow (E) and statistical analysis (F). ****P < .001. (G) FPKM of IRF8, BCL6, IRF4, PRDM1, BCL2L11, and BAX after treatment with romidepsin (R), talazoparib (T), and romidepsin plus talazoparib (R+T). MBL2 cells without any treatment were used as controls (C). RNASeq. *P < .05, **P < .01, ***P < .001, ****P < .0001. (H) Corresponding western blots of proteins after treatment with romidepsin (R), talazoparib (T), and romidepsin plus talazoparib (R+T). (I) Proposed schema of activation of apoptosis during the combination therapy with romidepsin and talazoparib.
Discussion
PARP-1 is an abundant nuclear enzyme that catalyzes poly(ADP-ribose)-ylation of target proteins, using nicotinamide adenine dinucleotide as a cofactor.24 For quite some time, PARP-1 was mostly seen as an enzyme that initiates DNA repair5,6 ; however, recent studies showed that PARP-1 poses many pleiotropic functions involved in epigenetic and transcriptional controls,25 regulates mRNA stability and decay,26 and has activities associated with oncogenic properties.27 Although PARP-1 can rarely be expressed in normal tissue, the level of its expression is particularly high in various cancers, such as leukemias, lymphomas, colorectal cancer, and especially breast and ovarian cancers.5,6 Previously, we showed that PARP-1 expression is increased in MF compared with healthy tissue.8 We also demonstrated in the past that PARP-1 expression in MF is increased in tumor stage compared with early-stage disease.8 In the present study, we have demonstrated that high PARP-1 expression on malignant lymphocytes correlates with worse OS in patients with MF. Similar observations were made recently in patients with acute myeloid leukemia, where high expression levels of PARP-1 were associated with worse OS and relapse-free survival.28
Originally indicated for BRCA-associated tumors and currently FDA approved for advanced ovarian cancer and metastatic breast cancer,9 PARPi are currently being explored for other malignancies such as head and neck squamous cell carcinoma,10,11 gastric cancer,12,13 osteosarcoma,14 and in chronic leukocytic leukemia.15,16 Talazoparib is a novel, selective inhibitor of PARP-1 and PARP-2 that has been shown to achieve antitumor response at lower concentrations than earlier generation PARPi.29 In marked contrast to PARP-1, loss of PARP-2 does not result in additional phenotypes in growth, development, or tumorigenesis in mice.30 We showed that MBL2 sensitivity to talazoparib was comparable with MDA-MB-436, BRCA1/2 mutant cells previously reported as being a highly sensitive cell line to talazoparib.23 The reason for the high IC50 values of talazoparib in MBL2 (3 logs over what one expects for a sensitive cell line) and MDA-MB-436 in our experiments was because of the different number of cells that we used for our assays. We used 7 × 104 cells per well in 96-well plates when Fejzo et al23 used 5 × 103 to 10 × 103 cells per well. Interestingly, the human lymphoma cell line HUT78 was more sensitive to talazoparib than MBL2, which provides some translational expectations. Thus, comparable MBL2 IC50 with MDA-MB-436, which is highly sensitive to talazoparib, breast cancer cell line, and even the higher sensitivity of human lymphoma cell line, HUT78, indicates clinical relevance of the current therapeutic doses and may be translated directly into the clinical trial.
We have shown that talazoparib arrested the cell cycle of lymphoma cells in G2/M because of upregulation of p53 and p21, which is consistent with previously published data obtained in breast cancer cell lines.31 PARP inhibition is not always sufficient to cause cell death,32 and the search for efficient combinations is ongoing. Previous studies in chronic lymphocytic leukemia showed that the activity of talazoparib is independent on the presence of WT p53 or complete loss of p53, indicating the importance of the other mechanism of action of PARPi in leukemia and lymphomas.15 PARP-1 is involved in DNA methylation by regulation of expression levels or protein activity of DNA methyltransferase-1. Abnormal DNA methylation is a characteristic feature of MF; thus, it will be reasonable to assume that MF patients might benefit from a combination of hypomethylating agents and PARPi. In this study, we have demonstrated the potentiation of the effect of PARP inhibition by HDACi. When talazoparib was combined with HDACi, romidepsin, the cytotoxic effect was synergized via downregulation of DNA-repair genes, FANCA, FANCD2, and TOPBP1 and stimulation of apoptosis via Blimp-1/Bax axis. Romidepsin increased the expression of IRF8 and BCL6, leading to upregulation of Blimp1 and Bax, whereas talazoparib upregulated Blimp-1 and BAX via upregulation IRF4 leading to cleavage of caspases 6 and 7. Thus, we have shown that the combination of PARPi with HDACi has a synergistic antilymphoma effect in vitro and in vivo, which warrants further investigation of this combination in clinical trials.
When PARPi was combined with HDACi in our experiments, the cytotoxic effect of that combination was mediated by the upregulation of Blimp-1 (PRDM1 gene), leading to apoptosis of lymphoma cells. PRDM1 is a transcriptional repressor, which has been shown to regulate the differentiation of B cells into antibody-secreting cells.33 The repression of MYC,34 SPIB, BCL6, ID3, and PAX5 is required for this function.35 Recently, PRDM1 expression has been detected in CD8+ and CD4+ T cells with the effector phenotype36,37 and in CD25+ regulatory T cells.37 Moreover, PRDM1 has been shown to regulate T-cell homeostasis36,37 and act as a tumor suppressor gene in natural killer cell malignancies.38 The baseline expression of PRDM1 was very low in our T-cell lymphoma cells, which is consistent with observation in natural killer cell lymphomas.38 It is possible that the mechanisms of PRDM1 inactivation are similar among those type of lymphoma, but further studies are needed to clarify this hypothesis.
In summary, the present results indicate the PARP-1 expression is upregulated in a cohort of patients with advanced MF whose disease has worse OS. PARP inhibition by talazoparib initiates an arrest of the cell cycle of lymphoma cells at G2/M and induces apoptosis.
For data sharing requests, e-mail the corresponding author, Oleg E. Akilov (akilovoe@upmc.edu).
RNAseq data were uploaded to the National Center for Biotechnology Information Gene Expression Omnibus database (accession number GSE157434).
Acknowledgments
The authors are grateful to Maria Aquafondata for excellent assistance with PARP-1 staining of skin samples from patients with mycosis fungoides.
This work was supported by funding from Pfizer Inc. (O.E.A.).
Authorship
Contribution: O.E.A. designed the research; O.K. performed the experiments; X.W. and S.T.H. provided the critical cell line and contributed to the data analysis; O.E.A. wrote the manuscript; and all authors read and approved the final draft.
Conflict-of-interest disclosure: O.E.A. is a recipient of a research grant from Pfizer Inc. that manufactures talazoparib. The remaining authors declare no competing financial interests.
Correspondence: Oleg E. Akilov, Department of Dermatology, University of Pittsburgh, 3708 Fifth Ave, 5th Floor, Suite 500.68, Pittsburgh, PA 15213; e-mail: akilovoe@upmc.edu.