

Key Points
Efficient clinical-scale correction of oxidase activity in X-linked CGD patient apheresis granulocytes is achieved by mRNA transfection.
Granulocyte-enriched apheresis products transfected with mRNA maintain high viability and functionality ex vivo and in vivo.

Abstract
Granulocytes from patients with chronic granulomatous disease (CGD) have dysfunctional phagocyte reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase that fails to generate sufficient antimicrobial reactive oxidative species. CGD patients with severe persistent fungal or bacterial infection who do not respond to antibiotic therapy may be given apheresis-derived allogeneic granulocyte transfusions from healthy volunteers to improve clearance of intractable infections. Allogeneic granulocyte donors are not HLA matched, so patients who receive the donor granulocyte products may develop anti-HLA alloimmunity. This not only precludes future use of allogeneic granulocytes in an alloimmunized CGD recipient, but increases the risk of graft failure of those recipients who go on to need an allogeneic bone marrow transplant. Here, we provide the first demonstration of efficient functional restoration of CGD patient apheresis granulocytes by messenger RNA (mRNA) electroporation using a scalable, Good Manufacturing Practice–compliant system to restore protein expression and NADPH oxidase function. Dose-escalating clinical-scale in vivo studies in a nonhuman primate model verify the feasibility, safety, and persistence in peripheral blood of infusions of mRNA-transfected autologous granulocyte-enriched apheresis cells, supporting this novel therapeutic approach as a potential nonalloimmunizing adjunct treatment of intractable infections in CGD patients.
Introduction
Chronic granulomatous disease (CGD) results from an inherited deficiency in production of antimicrobial reactive oxidative species (ROS) by granulocytes and monocytes.1,2 Patients with CGD suffer from recurrent severe bacterial and fungal infections,3,4 occurring at rates of 0.3 to 0.4 per year, that are the primary cause of morbidity and early mortality in CGD patients.5 The different genetic forms of CGD are caused by mutations in genes encoding any of the phagocyte reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase subunit proteins (gp91phox ∼70% of cases; p47phox ∼25%; p67phox ∼2.5%; p22phox ∼2.5%; and p40phox <1%).
Allogeneic donor hematopoietic stem cell (HSC) transplantation (HSCT) can provide definitive cure of CGD, and HSCT has been used in CGD as urgent salvage treatment of severe infections that have exhausted medical options.6 Alternatively, over the past 30 years, severely infected patients with CGD not responding to prolonged antibiotic therapy have in some centers been treated with allogeneic granulocyte transfusions with a reported 80% rate of improved disease burden.7 However, it is not practical to locate HLA-matched granulocyte apheresis donors, and infusion of non–HLA-matched allogeneic granulocytes can induce an alloimmune response, which not only precludes further use of apheresis granulocytes but also increases the risk of graft rejection by patients who proceed to allogeneic HSCT.8-10 Increasing concern for the alloimmunization risk of mismatched donor apheresis granulocyte transfusions has resulted in a significant decrease in its use as adjunct therapy for CGD infections. To address this problem of alloimmunity, we sought to develop a novel approach of repurposing patients’ autologous apheresis granulocytes by correcting them ex vivo so they might be fit to use for reinfusion in management of severe infections without inducing alloimmunization.
Recent improvements in the in vitro synthesis and quality control of messenger RNA (mRNA) that do not induce adverse cellular protective responses have raised the possibility of using such Good Manufacturing Practice (GMP)–grade mRNA for therapeutic purposes.11,12 Simultaneously, improvements in clinically compliant electroporators have opened new avenues for therapeutic intervention. For example, primary dendritic cells, T cells, B cells, and blood mononuclear leukocytes can be electroporation (EP) transfected with mRNAs to induce production of T-cell receptors, chimeric antigen receptors, adhesion receptors, and signal transducers for clinical use without adversely affecting viability or function.11,12 Moreover, we and others have demonstrated efficient in vitro delivery of mRNA to human CD34+ HSCs with resultant high viability and function using EP in the context of gene editing.13,14
We describe in this report the development of a novel clinically scalable approach to induce high rates of protein expression from exogenous mRNA electroporated into granulocyte-enriched apheresis cells from granulocyte colony-stimulating factor (G-CSF; filgrastim) treated human volunteers and nonhuman primates. More specifically, we demonstrate efficient in vitro functional restoration of phagocyte NADPH oxidase in treated cells from patients with 2 of the 4 genetic forms of CGD (X-linked CGD [X-CGD] deficiency of gp91phox; autosomal recessive (AR) p47phox-CGD deficiency of p47phox). We also used the GMP-compliant clinically scalable EP system to conduct preclinical Investigational New Drug (IND)–enabling studies in nonhuman primates, where infusions with escalating doses of enhanced green fluorescence protein (eGFP) mRNA-electroporated autologous apheresis cells were shown to be safe, well tolerated, and maintained viably expressing eGFP in peripheral blood.
Materials and methods
Research subjects
Peripheral blood apheresis products were collected from healthy volunteers (HVs) and X-CGD patients in the Department of Transfusion Medicine of the National Institutes of Health (NIH) Clinical Center after written informed consent following the Declaration of Helsinki per NIH Institutional Review Board–approved protocols 05-I-0213 and 94-I-0073. Most subjects undergoing apheresis received 5 daily injections of G-CSF at 10 to 16 µg/kg. Some also receive 1 dose of plerixafor the evening before collection if mobilized CD34+ HSCs were the main purpose for the collection unrelated to our granulocyte studies. However, some CGD patients underwent apheresis collection following a single dose of G-CSF plus 1 dose of dexamethasone mobilization to only collect products enriched in granulocytes. The granulocytes in apheresis products collected following G-CSF administration differ from the granulocyte populations found in the circulation of subjects not treated with G-CSF in that there are a greater proportion of immature granulocytes including band and other immature forms. Apheresis products with <60% granulocytes underwent elutriation to enrich for granulocytes, referred to as granulocyte-enriched products, or Grans. For some studies, small samples of peripheral blood were obtained from healthy volunteers or patients with CGD (protocol 05-I-0213).
Transfection with mRNA
The granulocyte-enriched apheresis products underwent elutriation to assure that >60% of granulocyte were electroporated, then washed 3 times with Hyclone EP buffer (MaxCyte) (+1% human serum albumin), then resuspended in EP buffer at 5 × 108/mL to 7.5 × 108/mL or as indicated. Other cells present in the starting cell mix included monocytes and lymphocytes (T, B, natural killer), where granulocytes/monocytes comprised >89% and lymphocytes <2.5%. The cells were mixed well with mRNA, then transfected in the appropriately sized Processing Assembly (PA; OC-100/400) per the proprietary program for granulocytes (GMP-compliant MaxCyte GT System). Post-EP, cells were incubated for 20 minutes at 37°C, then cultured at 5 × 106/mL to 7 × 106/mL. Cells were analyzed for viability, protein expression, and NADPH oxidase function at indicated times. Cells that were infused into mice or primates were cultured overnight in RPMI1640 supplemented with fetal bovine serum (10%) at 37°C. All mRNA were designed and produced by CELLSCRIPT, LLC (Madison, WI) by a proprietary processes designed to reduce immune responses against the exogenous mRNA. More specifically, mRNAs were prepared from a DNA template having a human codon-optimized open reading frame, globin 5′ and 3′ untranslated regions, 5′ cap with a cap1 or cap0 structure (respectively, for the p47phox mRNA vs gp91phox mRNA), pseudouridine in place of uridine and a 3′ poly(A) tail with ≥150 A’s. Although 4 noncoding structure variants of the mRNA were produced by CELLSCRIPT and assessed in many preliminary EP transfection mRNA-dosing experiments, no performance differences were noted between these different preparations as assessed by 2-way analysis of variance (ANOVA; F = 0.546; P = .654).
Flow cytometry
Intracellular gp91phox stain, p47phox stain and dihydrorhodamine 123 (DHR; Sigma-Aldrich) were performed as previously described,15,16 with phycoerythrin- or allophycocyanin-conjugated anti-human CD45, and human gp91phox or p47phox expression with murine monoclonal antibody 7D5 (MBL) or p47phox antibody, respectively, followed by fluorescein isothiocyanate–conjugated goat anti-mouse immunoglobulin G (IgG) antibody (BD Biosciences), and acquired using FACSCanto (Argon laser; Becton Dickinson) and analyses performed using FlowJo version 9.9.6 software for macOS (TreeStar Inc).
In vitro Aspergillus fumigatus hyphal damage experiments
Using a previously described colorimetric assay,17 the percentage of antifungal activity was measured by damage to opsonized hyphae. Hyphae grown from Aspergillus conidia (but additionally opsonized with 20% human serum) were incubated overnight at a 1:16 ratio with the test granulocyte-enriched apheresis cells from healthy volunteers, and X-CGD patient cells (naive or treated). Hyphal viability was determined as a function of mitochondrial redox potential using a colorimetric assay that assesses the reduction of 2,3-bis[2-methyloxy-4-nitro-5-[(sulfenylamino) carbonyl]-2H-tetrazolium-5-carboxanilide] (XTT) to formazan. Specifically, the hyphae were incubated with phosphate-buffered saline containing XTT (0.5 mg/mL) and coenzyme Q (40 µg/mL) for 1 hour at 37°C; XTT reduction was measured by recording the background subtracted absorbance value at 450 nm minus background absorbance recorded at 650 nm. The damage to hyphae is expressed as the percentage of antifungal activity per the following formula (in which OD represents optical density and G represents Grans):
% antifungal activity = [1 − (ODAf+G − ODG)/(ODAf − ODBlank)] × 100
ODAf+G is (OD450 − OD650) of wells containing A fumigatus hyphae with G.
ODG is (OD450 − OD650) of wells containing G.
ODAf is (OD450 − OD650) of wells containing A fumigatus hyphae alone.
ODblank is (OD450 − OD650) of wells containing media alone.
Assays for each condition were performed (n = 6).
Mouse studies
Adult NOD.SCIDγ-Prkdcscid Il2rgtm1Wjl/SzJ mice (The Jackson Laboratory) under National Institute of Allergy and Infectious Diseases (NIAID) Institutional Animal Care and Use Committee (ACUC)–approved protocol LCIM 1E were injected with either 100, 200, or 300 million cells from the healthy volunteers or CGD patient apheresis granulocyte preparations (grans) per animal (n = 20). Following injection, mice were analyzed for the presence of human cells in the peripheral blood or in organs (harvest at euthanasia at day 3) or monitored for survival and side effects over 2 weeks.
Nonhuman primate studies
Rhesus macaque nonhuman primate (NHP) safety studies (NHLBI ACUC Protocol H-0136R3) evaluated the safety and persistence in circulation for 4 to 5 days following the infusion of increasing numbers of eGFP-mRNA–transfected autologous leukocyte apheresis cells. As only 2 animals were used (though with 1 animal undergoing apheresis and infusion of transfected cells multiple times), a complete descriptive analysis of the data were provided instead of summary statistics. These 2 NHPs had previously received lentivector eGFP-transduced autologous HSCs and stably express eGFP+ in a low percentage of circulating blood cells that ensured immune tolerance to the eGFP, but still allowed assessment of circulating eGFP-expressing cells resulting from infusions of the eGFP-mRNA EP autologous apheresis leukocytes. The NHP received 1 dose (15 µg/kg) of G-CSF plus 1 mg/kg dexamethasone 12 to 24 hours prior to apheresis. Leukocyte apheresis products were collected and, after eGFP mRNA transfection, reinfused into the NHPs.
In the NHP that received 3 collections and transfusions, cells were administered at 1 × 107 cells per kilogram initially, and then 1 × 108 cells per kilogram, and then 5 × 108 cells per kilogram. Cells were administered at 1 × 107/mL to 5 × 107/mL in saline, infused over 15 to 20 minutes. Chest radiographs were taken prior to and following cell administration. Vital signs, including blood pressure, were monitored. Blood was drawn prior to, 30 minutes, 1 hour, 24 hours, then at 2, 3, 4 and ± 5 days following infusion to monitor eGFP expression in peripheral blood. Both animals were retained for long-term assessment of general status.
NanoString analysis
RNA extracted from granulocytes (Qiagen RNeasy Plus Mini kit) was evaluated for expression of 255 inflammation-related and housekeeping genes (GX Human Inflammation kit; NanoString XT CodeSet) per the manufacturer’s instructions (NanoString Technologies) and read using the nCounter digital analyzer (CCR Genomics Core, National Cancer Institute [NCI], NIH). Raw data were normalized using nSolver Analysis software 4.0.
Statistical analysis
All data are presented with Prism 7 GraphPad software (version 7.0d). For statistical analysis of p47phox expression following EP, we treated time and dosage as independent categorical variables, and p47phox expression as the response variable; the 2-way ANOVA was used to assess differences between treatment groups while also accounting for variation in expression over time (http://www.biostathandbook.com/twowayanova.html). The following assumptions are made, that Time and Dosage act independently of each other, and that each data cell is normally distributed, with equal variances between cells.
Results
Optimization of the transfection protocol with eGFP mRNA
Using eGFP mRNA, we determined optimum conditions for EP transfection in healthy volunteers’ granulocyte-enriched apheresis products henceforth referred to as “Grans.” We observed rapid high level eGFP expression in >90% of cells both at 2 and 24 hours in culture post-EP (Figure 1A). Incubating the cells with eGFP mRNA at room temperature for 5 and 10 minutes prior to EP progressively decreased eGFP expression to the no-expression baseline (Figure 1B), whereas incubation on ice for up to 10 minutes with or without red blood cell lysis maintained ≥90% eGFP-expressing cells at 24 hours with no significant impact on viability (Figure 1B). However, this same approach when applied to granulocytes isolated from peripheral blood using Percoll-based density gradient or Ficoll-Paque premium density gradient did not achieve eGFP expression.
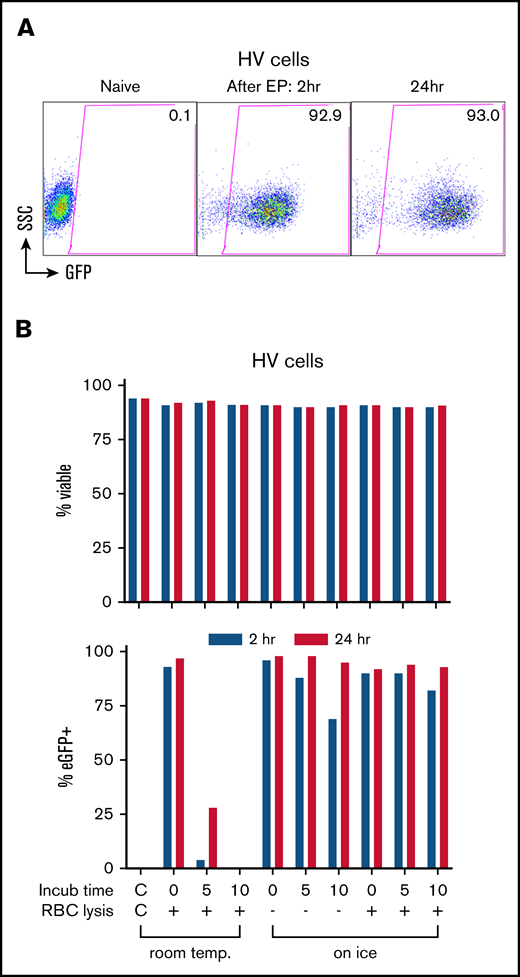
Transfection of leukapheresis cells by EP with mRNA. (A) EP transfection of the HV granulocyte-rich apheresis product with eGFP mRNA. Shown are dot plot graphs of FACS analysis of eGFP expression in naive cells and at 2 and 24 hours after transfection. Percentage of eGFP+ is indicated in the gated areas. (B) Optimization of transfection conditions assessing time (minutes) and temperature (temp.; room temp vs water ice) of incubation with eGFP mRNA before EP, and the effect of a red cell lysis step (+) before EP on the viability (top panel) and transfection efficiency (bottom panel) at 2 hours (blue) and 24 hours (red) after EP. The leftmost bars in graphs show the non-EP control (C/C). Incub, incubation; RBC, red blood cell; SSC, side scatter.
Transfection of leukapheresis cells by EP with mRNA. (A) EP transfection of the HV granulocyte-rich apheresis product with eGFP mRNA. Shown are dot plot graphs of FACS analysis of eGFP expression in naive cells and at 2 and 24 hours after transfection. Percentage of eGFP+ is indicated in the gated areas. (B) Optimization of transfection conditions assessing time (minutes) and temperature (temp.; room temp vs water ice) of incubation with eGFP mRNA before EP, and the effect of a red cell lysis step (+) before EP on the viability (top panel) and transfection efficiency (bottom panel) at 2 hours (blue) and 24 hours (red) after EP. The leftmost bars in graphs show the non-EP control (C/C). Incub, incubation; RBC, red blood cell; SSC, side scatter.
Transfection with p47phox mRNA to correct AR p47phox-CGD apheresis granulocytes
We next assessed effect of mRNA concentrations and cell densities during EP using p47phox mRNA to correct Grans from 3 patients with AR p47phox-deficient CGD. EP using p47phox mRNA concentrations up to 400 µg/mL (Figure 2A) or at cell concentrations up to 1 × 109/mL during EP had minimal impact on cell viability (Figure 2B) compared with non-EP control. Next, we assessed restoration of p47phox protein expression and NADPH oxidase activity by flow cytometry post-EP (Figure 2C) and demonstrate the kinetics of restoration of p47phox expression (Figure 2D) and NADPH oxidase activity (Figure 2E) following EP at 3 mRNA concentrations (200, 300, and 400 µg/mL) over time up to 120 hours. Of note, although the p47phox expression was measurable in only 10% to 35% of cells by 2 hours (Figure 2D), NADPH oxidase activity was ∼90% restored by then and was maintained in ∼75% (of viable cells) out to 120 hours (Figure 2E). The p47phox expression peaked at ∼84% of cells at 4 hours and significantly decreased by 72 hours (Figure 2D). Using mRNA concentration at 400 μg/mL achieved the highest percentage of cells expressing p47phox and NADPH oxidase activity (Figure 2D-E) for different cell doses evaluated. Despite the low percentage of p47phox protein-expressing cells by day 5 (120 hours), NADPH oxidase activity was maintained in ∼75% of the remaining viable cells at half the maximum level per cell even out to 5 days. This indicates that the relatively low amounts of p47phox protein in remaining viable cells at 5 days were sufficient to support significant per cell correction of NADPH oxidase function for almost the duration of the neutrophil lifespan between 5 and 90 hours in circulation.15,18 It is also possible that monocytes in the product were electroporated and contribute to the extended NADPH oxidase activity.
![Transfection with p47phoxmRNA to correct AR CGD granulocytes. Correction of AR CGD p47phox-deficient patient cells with p47phox mRNA. Granulocyte-enriched apheresis products from 3 different AR p47phox-CGD patients were transfected by EP with p47phox mRNA for each condition shown. (A) Effect of p47phox mRNA concentrations at EP on cell viability up to 120 hours post-EP at a constant cell number of 5 × 108 cells per milliliter in EP mix (n = 3 ± standard deviation [SD]; control = non-EP). (B) Effects of varying cell concentrations in EP mix on cell viability up to 120 hours post-EP at 200 or 300 µg of mRNA per milliliter. (C) Representative FACS dot plot analyses of granulocyte-rich apheresis product at times indicated after EP treatment of p47phox-deficient AR CGD patient 5 × 108 cells per milliliter with 400 µg of mRNA per milliliter in EP mix (treated) or non-EP (not treated) control. Top left panel of side scatter (SSC) by forward scatter (FSC) shows gate that captures both mature and immature granulocytes in the granulocyte-rich apheresis product used for analyses shown in the other panels. For the not treated and treated 4-hour and 24-hour groups, the top panels show p47phox expression and the bottom panels show phorbol 12-myristate 13-acetate (PMA)–stimulated increase of DHR fluorescence as a measure of oxidase activity, where the percentage of cells in the positive gates is shown at the top of each panel and the mean fluorescence intensity (MFI) as a measure of expression per positive cell is shown at the bottom of each panel. (D) The kinetics of p47phox protein expression shown as a percentage of p47phox+ cells over 120 hours in culture post-EP. For each sample, 5 × 108 cells per milliliter were treated at the indicated p47phox mRNA concentrations in the EP mix (n = 3 ± SD; control = non-EP). (E) The kinetics of NADPH oxidase activity shown as percentage of DHR+ cells over 120 hours in culture post-EP for the same preparations as in panel D (n = 3 ± SD; control = non-EP). Using 2-way ANOVA analysis, no effect on viability due to cell concentrations (F = 0.241, P = .627) or p47phox mRNA concentrations (F = 1.252, P = .307) was observed.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/23/10.1182_bloodadvances.2020003224/4/m_advancesadv2020003224f2.png?Expires=1769176957&Signature=GZm6pyOiD~qlxIpXHy3YI-IeQ6XePJRxLyEHI8SobPFFbZjzcHr7Zxsx7D-Sc~aFXfizxdT4NFfyQyv~9PWnpM7GsEljJK3Neb9gLaWd8I1LiyXRApDkrfdgZgBfq0oOCJcVfL~hDtYwXvYi7eG6E4TfNOYoB~y6wozJt2LJM533-5yhkSZt0kXy7g39u3WPGMuVj3rUtuwNIOS~ipzNNYFmklihGdc2yraRwsH7g6glHaNEJME-ELdeJzLs~1fOIHOYi2gZ4kxKD4mn~xKfx-l6gnVkicuxbPbblZpzYIcbte3olk~sx9BvtFiqXWpan0ttg7ibQJuejH3PximDHg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Transfection with p47phoxmRNA to correct AR CGD granulocytes. Correction of AR CGD p47phox-deficient patient cells with p47phox mRNA. Granulocyte-enriched apheresis products from 3 different AR p47phox-CGD patients were transfected by EP with p47phox mRNA for each condition shown. (A) Effect of p47phox mRNA concentrations at EP on cell viability up to 120 hours post-EP at a constant cell number of 5 × 108 cells per milliliter in EP mix (n = 3 ± standard deviation [SD]; control = non-EP). (B) Effects of varying cell concentrations in EP mix on cell viability up to 120 hours post-EP at 200 or 300 µg of mRNA per milliliter. (C) Representative FACS dot plot analyses of granulocyte-rich apheresis product at times indicated after EP treatment of p47phox-deficient AR CGD patient 5 × 108 cells per milliliter with 400 µg of mRNA per milliliter in EP mix (treated) or non-EP (not treated) control. Top left panel of side scatter (SSC) by forward scatter (FSC) shows gate that captures both mature and immature granulocytes in the granulocyte-rich apheresis product used for analyses shown in the other panels. For the not treated and treated 4-hour and 24-hour groups, the top panels show p47phox expression and the bottom panels show phorbol 12-myristate 13-acetate (PMA)–stimulated increase of DHR fluorescence as a measure of oxidase activity, where the percentage of cells in the positive gates is shown at the top of each panel and the mean fluorescence intensity (MFI) as a measure of expression per positive cell is shown at the bottom of each panel. (D) The kinetics of p47phox protein expression shown as a percentage of p47phox+ cells over 120 hours in culture post-EP. For each sample, 5 × 108 cells per milliliter were treated at the indicated p47phox mRNA concentrations in the EP mix (n = 3 ± SD; control = non-EP). (E) The kinetics of NADPH oxidase activity shown as percentage of DHR+ cells over 120 hours in culture post-EP for the same preparations as in panel D (n = 3 ± SD; control = non-EP). Using 2-way ANOVA analysis, no effect on viability due to cell concentrations (F = 0.241, P = .627) or p47phox mRNA concentrations (F = 1.252, P = .307) was observed.
Transfection with p47phoxmRNA to correct AR CGD granulocytes. Correction of AR CGD p47phox-deficient patient cells with p47phox mRNA. Granulocyte-enriched apheresis products from 3 different AR p47phox-CGD patients were transfected by EP with p47phox mRNA for each condition shown. (A) Effect of p47phox mRNA concentrations at EP on cell viability up to 120 hours post-EP at a constant cell number of 5 × 108 cells per milliliter in EP mix (n = 3 ± standard deviation [SD]; control = non-EP). (B) Effects of varying cell concentrations in EP mix on cell viability up to 120 hours post-EP at 200 or 300 µg of mRNA per milliliter. (C) Representative FACS dot plot analyses of granulocyte-rich apheresis product at times indicated after EP treatment of p47phox-deficient AR CGD patient 5 × 108 cells per milliliter with 400 µg of mRNA per milliliter in EP mix (treated) or non-EP (not treated) control. Top left panel of side scatter (SSC) by forward scatter (FSC) shows gate that captures both mature and immature granulocytes in the granulocyte-rich apheresis product used for analyses shown in the other panels. For the not treated and treated 4-hour and 24-hour groups, the top panels show p47phox expression and the bottom panels show phorbol 12-myristate 13-acetate (PMA)–stimulated increase of DHR fluorescence as a measure of oxidase activity, where the percentage of cells in the positive gates is shown at the top of each panel and the mean fluorescence intensity (MFI) as a measure of expression per positive cell is shown at the bottom of each panel. (D) The kinetics of p47phox protein expression shown as a percentage of p47phox+ cells over 120 hours in culture post-EP. For each sample, 5 × 108 cells per milliliter were treated at the indicated p47phox mRNA concentrations in the EP mix (n = 3 ± SD; control = non-EP). (E) The kinetics of NADPH oxidase activity shown as percentage of DHR+ cells over 120 hours in culture post-EP for the same preparations as in panel D (n = 3 ± SD; control = non-EP). Using 2-way ANOVA analysis, no effect on viability due to cell concentrations (F = 0.241, P = .627) or p47phox mRNA concentrations (F = 1.252, P = .307) was observed.
Transfection with gp91phox mRNA to correct X-CGD granulocytes
We next applied the optimized EP conditions determined using eGFP and p47phox mRNA to granulocyte-enriched apheresis products from X-CGD patients (n = 3). At 24 hours following EP with gp91phox mRNA (400 µg/mL), ∼80% of the flow dot plot scattergram-gated granulocytes expressed gp91phox protein (Figure 3A top panels), and >80% of treated granulocytes were DHR+ (Figure 3A bottom panel). Of note, the kinetics of the expression of gp91phox, a transmembrane subunit, differed from that of p47phox, a cytosolic protein, in that the protein expression was sustained at a much higher level (>60%) for over 100 hours after the mRNA transfection (Figure 3B) with 50% DHR+ granulocytes (Figure 3C).

gp91 transfection with gp91phoxmRNA to correct X-CGD granulocytes. Correction of X-CGD gp91phox-deficient patient cells with gp91phox mRNA. (A) Representative FACS dot plot analyses of granulocyte-rich apheresis product at 4 hours and 24 hours after EP treatment of gp91phox -deficient X-CGD patient 5 × 108 cells per milliliter with 400 µg of mRNA per milliliter in EP mix (treated) or no-EP (not treated) control. The panels are arranged in the same order and correspond to the same analyses as described in the figure legend for Figure 2C and show expression of gp91phox protein and the PMA-stimulated NADPH oxidase activity (as increase in DHR fluorescence). (B) The kinetics of gp91phox protein expression shown as percentage of gp91phox+ cells over 120 hours in culture post-EP with 5 × 108 cells per milliliter with 400 µg/mL gp91phox mRNA (n = 3 ± SD; control = non-EP). (C) The kinetics of NADPH oxidase activity shown as percentage of DHR+ cells over 120 hours in culture post-EP for the same preparations as in panel B (n = 3 ± SD; control = non-EP).
gp91 transfection with gp91phoxmRNA to correct X-CGD granulocytes. Correction of X-CGD gp91phox-deficient patient cells with gp91phox mRNA. (A) Representative FACS dot plot analyses of granulocyte-rich apheresis product at 4 hours and 24 hours after EP treatment of gp91phox -deficient X-CGD patient 5 × 108 cells per milliliter with 400 µg of mRNA per milliliter in EP mix (treated) or no-EP (not treated) control. The panels are arranged in the same order and correspond to the same analyses as described in the figure legend for Figure 2C and show expression of gp91phox protein and the PMA-stimulated NADPH oxidase activity (as increase in DHR fluorescence). (B) The kinetics of gp91phox protein expression shown as percentage of gp91phox+ cells over 120 hours in culture post-EP with 5 × 108 cells per milliliter with 400 µg/mL gp91phox mRNA (n = 3 ± SD; control = non-EP). (C) The kinetics of NADPH oxidase activity shown as percentage of DHR+ cells over 120 hours in culture post-EP for the same preparations as in panel B (n = 3 ± SD; control = non-EP).
In vitro phagocytic and antimicrobial activity of the granulocyte-rich apheresis product after mRNA EP transfection correction
We assessed maintenance of phagocytosis function by the p47phox-deficient AR CGD patient-derived granulocyte-rich apheresis product either unmanipulated (non-EP) or after p47phox mRNA EP transfection correction of oxidase activity over 48 hours by assessing phagocytosis of inactivated fluorescent particles of Staphylococcus aureus (Figure 4A).
![In vitro analysis of phagocytosis, antimicrobial activity, and gene-expression profiles of mRNA-electroporated granulocytes. (A) Phagocytosis capacity after incubation of p47phox-deficient AR CGD granulocyte-rich apheresis cells either non-EP or p47phox mRNA EP transfection corrected with inactivated AF488 fluorescent-labeled particles of S aureus (multiplicity of infection [MOI], 5) (n = 1-4). (B) Hyphal damage to A fumigatus expressed as the percentage of antifungal activity by HV granulocyte-rich apheresis product non-EP control or eGFP mRNA transfection control compared with X-CGD patient granulocyte-rich apheresis product non-EP (naive), eGFP mRNA EP transfection control, or gp91phox mRNA transfection corrected (n = 6 replicates for each condition). ANOVA test with multiple comparisons; *P < .05. (C) Gene-expression profiles in mRNA-transfected granulocytes at 24 hours post-EP. Log2 fold-change of expression for genes related to the response to exogenous mRNA for EP mRNA samples relative to naive samples for each 3 CGD patients (P1-3). Mean of 2 experimental replicates shown for P1 and P3 and a single analysis for P2. Genes with absolute log2 fold-change value ≥2 are formally defined as true positives. ns, nonsignificant.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/23/10.1182_bloodadvances.2020003224/4/m_advancesadv2020003224f4.png?Expires=1769176957&Signature=ZdG7NuBTN2n1C3DH7AUzJKywTXQauX7OA82Pl15dzTww3B-hZ5WaeaAD9gkairahX1oxSHT862vJ2~qNd8T~-gcTDNYMKwPkMvfR5yXQD37RAeZeW~eTGKUDXbzU4LN77H6FboJuxL7OpmUEd-S2~PSBH~CopreYcf6GVp83RfcobwAp1osDAj3E8cfD2sgE8-kLNpYT9bnB8KPqTX~k-7ajTyfS~2Lov43DbLO-PRRGyuJx1CePhnolfMTxjmxdH7ePu6DS~Z39V1jCM8w4~k9PYMsMdHHHRgtYEgYgquJdhzdkxC7Uf11uw8fzE-0um3QOxOPpOwwXHssqmVXxfg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
In vitro analysis of phagocytosis, antimicrobial activity, and gene-expression profiles of mRNA-electroporated granulocytes. (A) Phagocytosis capacity after incubation of p47phox-deficient AR CGD granulocyte-rich apheresis cells either non-EP or p47phox mRNA EP transfection corrected with inactivated AF488 fluorescent-labeled particles of S aureus (multiplicity of infection [MOI], 5) (n = 1-4). (B) Hyphal damage to A fumigatus expressed as the percentage of antifungal activity by HV granulocyte-rich apheresis product non-EP control or eGFP mRNA transfection control compared with X-CGD patient granulocyte-rich apheresis product non-EP (naive), eGFP mRNA EP transfection control, or gp91phox mRNA transfection corrected (n = 6 replicates for each condition). ANOVA test with multiple comparisons; *P < .05. (C) Gene-expression profiles in mRNA-transfected granulocytes at 24 hours post-EP. Log2 fold-change of expression for genes related to the response to exogenous mRNA for EP mRNA samples relative to naive samples for each 3 CGD patients (P1-3). Mean of 2 experimental replicates shown for P1 and P3 and a single analysis for P2. Genes with absolute log2 fold-change value ≥2 are formally defined as true positives. ns, nonsignificant.
In vitro analysis of phagocytosis, antimicrobial activity, and gene-expression profiles of mRNA-electroporated granulocytes. (A) Phagocytosis capacity after incubation of p47phox-deficient AR CGD granulocyte-rich apheresis cells either non-EP or p47phox mRNA EP transfection corrected with inactivated AF488 fluorescent-labeled particles of S aureus (multiplicity of infection [MOI], 5) (n = 1-4). (B) Hyphal damage to A fumigatus expressed as the percentage of antifungal activity by HV granulocyte-rich apheresis product non-EP control or eGFP mRNA transfection control compared with X-CGD patient granulocyte-rich apheresis product non-EP (naive), eGFP mRNA EP transfection control, or gp91phox mRNA transfection corrected (n = 6 replicates for each condition). ANOVA test with multiple comparisons; *P < .05. (C) Gene-expression profiles in mRNA-transfected granulocytes at 24 hours post-EP. Log2 fold-change of expression for genes related to the response to exogenous mRNA for EP mRNA samples relative to naive samples for each 3 CGD patients (P1-3). Mean of 2 experimental replicates shown for P1 and P3 and a single analysis for P2. Genes with absolute log2 fold-change value ≥2 are formally defined as true positives. ns, nonsignificant.
Next, we evaluated for restoration of the ability to damage A fumigatus hyphae expressed as the percentage of antifungal activity by the gp91phox-deficient X-CGD patient-derived granulocyte-rich apheresis product after gp91phox mRNA EP transfection correction of oxidase activity. We compared these corrected cells to the no-EP-naive or eGFP mRNA EP-transfected gp91phox-deficient X-CGD patient-derived granulocyte-rich apheresis product, and the no-EP-naive or eGFP mRNA EP-transfected healthy volunteer-derived granulocyte-rich apheresis product. Corrected gp91phox-deficient X-CGD patient-derived granulocyte-rich apheresis product achieved a significant (P < .05) increase in the percentage of antifungal activity (mean, 20.34%) compared with uncorrected X-CGD controls (11.92% for naive X-CGD and 12.85% for eGFP-EP X-CGD) and was similar to the the percentage of antifungal activity of the HV non-EP control (11.92%) (P < .05, ANOVA test with multiple comparisons) (Figure 4B).
Evaluation of innate immune responses to mRNA EP in granulocytes
In addition to determining viability at the cellular level, we assessed endogenous cellular protective or inflammatory responses to the exogenous mRNA and EP. Exogenous nucleic acid may trigger pathogen pattern recognition receptors (PRRs), including the Toll-like receptors (TLRs; TLR3, TLR7, and TLR8) and retinoic-acid-inducible gene 1 (RIG-1)-like receptor activation of NF-κB, interferon regulatory factors (IRFs), type I interferon (IFN)–responsive genes and proinflammatory cytokines.16,19,20 The transcriptional status of 249 inflammatory-related genes in granulocytes at 24 hours post-EP was assessed using NanoString technology.21 We assessed granulocyte-rich apheresis products from 3 different p47phox CGD patients. Although the fine details of the transcriptional profile were unique to each patient, this was minimally impacted by the EP transfection with mRNA. More specifically, Figure 4C shows the quantitative assessment of the change from naive (no exposure to mRNA or EP) as compared with 1 replicate (P2) or the mean of 2 replicate (P1 and P3) mRNA NanoString analyses of mRNA EP transfection treatment each of the 30 key cellular response genes in the samples from 3 different patients. With respect to genes involved in the innate immune response pathway in response to the exogenous mRNA, there were minimal changes in expression of NFKB1 and IRF3 genes (Figure 4C). However, there was a pattern of increased variability (upregulation in P2 and P3 but downregulation in P1) exceeding the 2× log2 change in IFN-inducible oligoadenylate synthetase (OAS) enzyme genes (OASL and OAS2) and other IFN-inducible genes (MX1, IFI44, IFIT1, IFIT2, IFIT3). But taking into account all of the data for all 3 patients, even these changes were not statistically significant. Interestingly, there was no significant upregulation or downregulation of proinflammatory cytokine genes (IL1B, IL6, IL10, IL12, TGFB, and TNF) in the 3 patients’ granulocyte-rich apheresis products induced by the mRNA EP transfection. These data suggest that the mRNA transfection has no major impact on the inflammatory status of corrected granulocytes up to the 24-hour time tested.
Infusion of electroporated granulocyte-enriched apheresis cells into mice
To assess whether mRNA-electroporated apheresis cells could be infused safely and to evaluate the distribution and the kinetics of the infused cells, apheresis cells from a healthy volunteer electroporated with eGFP mRNA were injected into adult immunodeficient mice (n = 20) (Figure 5A-B). Due to animal care and safety precautions, peripheral blood (PB) from the animals was assessed at 24 hours postinjection and some animals were euthanized at 3 days for analysis of PB and multiple organs (liver, spleen, lungs) for the presence of eGFP-expressing human cells. Surprisingly, eGFP+ human cells were still detected at 1% to 2% in PB at 3 days following infusion (Figure 5A). Significant percentages of eGFP+ human cells were also extracted from lungs, liver, and spleen (Figure 5B). With the exception of mice euthanized for tissue evaluation for eGFP+ human cells, all remaining mice where monitored for up to 2 weeks after cell injection. It is important to note that no death or adverse effects were reported in any of the animals injected (n = 20) despite the high dose of cells injected (300 × 106 cells per 30-g mouse equivalent to a 1 × 1010 cells per kilogram dose), providing additional evidence for lack of adverse effects from the in vivo administration into mice of very high numbers per kilogram of mRNA EP-transfected Grans. This number of mRNA EP-transfected Grans per mice exceeds by almost twofold the maximum dose of a standard clinical granulocyte transfusion product given to patients.

In vivo persistence in blood and egress to tissues sites by mRNA EP-transfected granulocyte-rich apheresis product cells. Infusion of apheresis cells into mice. High doses of GFP mRNA-transfected granulocytes (up to 300 × 106 cells per mouse) were IV injected into mice at 24 hours post-EP. GFP expression (B) was followed in the human CD45+ cells (A) after 24 hours in the PB and after 72 hours in the PB (n = 3-5 animals), liver, lungs, and spleen (n = 1).
In vivo persistence in blood and egress to tissues sites by mRNA EP-transfected granulocyte-rich apheresis product cells. Infusion of apheresis cells into mice. High doses of GFP mRNA-transfected granulocytes (up to 300 × 106 cells per mouse) were IV injected into mice at 24 hours post-EP. GFP expression (B) was followed in the human CD45+ cells (A) after 24 hours in the PB and after 72 hours in the PB (n = 3-5 animals), liver, lungs, and spleen (n = 1).
Preclinical dose-escalation studies in NHPs
Next, we evaluated the safety of infusing mRNA-transfected Grans in NHPs (Figure 6) that permitted monitoring of vital signs and radiographs for determination of adverse events. Because there are no rhesus models for CGD, we chose eGFP mRNA for transfection into rhesus apheresis cells and then reinfusing the autologous transfected cells back into the donor macaque. To minimize immunogenic responses to foreign eGFP, we chose 2 animals (ZG21 and ZH32) that had previously received integrating vector eGFP-transduced autologous stem cells in an unrelated study (NHLBI ACUC–approved protocol H-0136R3)22 over 4 years prior to the current study. These 2 animals had established stable, low-level percentages of eGFP+ bright cells that varied between 0.2% and 3.0% of total cells over several years. The 8- to 9-kg animals were mobilized with 15 µg of G-CSF per kilogram subcutaneously and 1 mg of dexamethasone per kilogram intramuscularly administered ∼12 hours prior to the apheresis to enhance granulocyte production. A mobilization/apheresis cycle was performed on 3 occasions for ZG21 and once for ZH32 to collect 1 × 107/kg, 1 × 108/kg, 5 × 108/kg, and 5 × 108/kg, respectively. Following EP with eGFP mRNA, the apheresed cells rested overnight before reinfusion into the same animal. Following cell infusion, a chest radiograph was taken, and vital signs were monitored. PB was drawn prior to and at 5 or 30 minutes, 1 hour, 24 hours, and then daily up to 4 days (ZG21) or 5 days (ZH32) following infusions to evaluate eGFP expression in circulating blood cells.

Preclinical dose-escalation studies in NHPs. Studies in NHPs (rhesus macaque) injected with transfected autologous apheresis cells. (A) FACS analysis of GFP expression in apheresis products from ZG21 or ZH32 “treated” with GFP mRNA transfection or “untreated.” Side scatter (SSC) by forward scatter (FSC) panel indicates gating for panels to the right. Right panels measure GFP expression in the CD18+ granulocyte/monocyte populations (percentage of cells indicated). Baseline GFP-bright clusters are evident in “untreated” panels. (B) FACS analysis of GFP expression in PB from ZG21 or ZH32 “preinfusion” at 1 or 2 days postinfusion of 1 × 108 transfected apheresis cells. SSC by FSC dot plot indicates gating enriched for granulocyte/monocyte population. Two boxed areas in each panel indicate baseline bright GFP+ cluster (right box) and less bright GFP+ cluster (left box) measuring GFP expression from GFP mRNA transfection. (C) FACS analysis over time after infusion of the less bright GFP cluster (left box value per panel B) in PB from ZG21 (red lines) or ZH32 (blue line) at 5 and 10 minutes, 1 hour, and daily as indicated after infusion of indicated number of autologous transfected apheresis cells.
Preclinical dose-escalation studies in NHPs. Studies in NHPs (rhesus macaque) injected with transfected autologous apheresis cells. (A) FACS analysis of GFP expression in apheresis products from ZG21 or ZH32 “treated” with GFP mRNA transfection or “untreated.” Side scatter (SSC) by forward scatter (FSC) panel indicates gating for panels to the right. Right panels measure GFP expression in the CD18+ granulocyte/monocyte populations (percentage of cells indicated). Baseline GFP-bright clusters are evident in “untreated” panels. (B) FACS analysis of GFP expression in PB from ZG21 or ZH32 “preinfusion” at 1 or 2 days postinfusion of 1 × 108 transfected apheresis cells. SSC by FSC dot plot indicates gating enriched for granulocyte/monocyte population. Two boxed areas in each panel indicate baseline bright GFP+ cluster (right box) and less bright GFP+ cluster (left box) measuring GFP expression from GFP mRNA transfection. (C) FACS analysis over time after infusion of the less bright GFP cluster (left box value per panel B) in PB from ZG21 (red lines) or ZH32 (blue line) at 5 and 10 minutes, 1 hour, and daily as indicated after infusion of indicated number of autologous transfected apheresis cells.
No adverse events were observed in the animals for 5 days postinfusion. Low percentages of background bright eGFP expression is most evident in the ex vivo “untreated” apheresis samples (Figure 6A) and in the “preinfusion” blood samples from the in vivo studies (Figure 6B). EP of the rhesus Grans was highly efficient, with ∼83% to 90% of cells in the CD18+ granulocyte/monocyte “treated” population in vitro expressing eGFP (Figure 6A). Significant percentages of dimmer eGFP (3.6% to 11%) expressing granulocytes due to the eGFP mRNA transfection (Figure 6A leftmost boxed area) were clearly distinguishable from the baseline bright eGFP expression (Figure 6A rightmost boxed area) in granulocytes observed in circulating blood granulocytes at 1 and 2 days after an infusion of the autologous transfected Grans (Figure 6B). A more detailed assessment of eGFP expression over time after each of 4 infusions (at 5 minutes, 30 minutes, 1 hour, and then daily up to 5 days) showed early detection of significant numbers of (3.8% to 10%) circulating transfection-related eGFP+ granulocytes at 5 minutes after the infusion, maintained or peaked at 24 hours, decreasing slightly to modestly at days 2 to 3, and still detectable at low levels on day 4 or 5 (Figure 6C). It is important to note that 1 of the animals (ZG21) received 3 collections and infusions of eGFP mRNA-transfected cells on separate occasions and tolerated all 3 collections and infusions without any problem.
Cell-manufacturing controls for IND
In support of a clinical protocol to treat patients with mRNA-corrected autologous peripheral blood granulocytes, 5 performance qualifications (3 healthy volunteers, 2 CGD patients) were performed under clinical conditions at the NIH Department of Transfusion Medicine using pharmaceutical-grade p47phox or gp91phox mRNA on respective CGD patient cells, or eGFP mRNA on healthy volunteers’ cells. All validation runs, including a full-scale dose of 3 × 109 cells (equivalent to a dose of 1 × 108 cells per kilogram for a hypothetical 30-kg pediatric patient), were successful in manufacturing products that fulfilled release criteria with high viability and transgene protein expression detected at a high level in a range of 74.7% to 91.9% inclusive of all of the Chemistry Manufacturing and Controls (CMC) clinical-scale manufacturing runs (Table 1).
Discussion
Dysfunctional immune cells are a hallmark of most primary immunodeficiencies. Although potentially curative, the presence of infection with associated inflammatory burden at the time of HSC/bone marrow transplant or gene therapy can significantly worsen outcomes.6 Here, we show for the first time that primary leukocytes collected by apheresis may be transiently corrected by mRNA transfection to restore sufficient protein expression for cellular activity using a GMP-compliant EP system. We demonstrated this correction of granulocytes in X-CGD and the p47phox-deficient autosomal CGD.
Patients with CGD are at high risk of invasive infections that may fail treatment despite prolonged aggressive medical therapy, requiring surgery or even allogeneic HSCT or autologous gene-modified HSC gene therapy under high-risk conditions.6 Granulocyte transfusions from healthy volunteers at doses of 0.2 × 108/kg to 5 × 108/kg have been used at 1 to 3 times weekly for 6 to 8 weeks for treatment of severe intractable infections in CGD patients for several decades.5,7,23-25 Although shown to improve control of infections in up to 75% of cases, they are associated with numerous adverse events including fevers, transfusion-related events, and most importantly, the development of anti-HLA antibodies in 29% to 80% of cases.7,9 Alloimmune responses can also decrease efficacy of subsequent granulocyte transfusions by reducing the circulating lifespan of donor granulocytes.7,9 An earlier report in CGD patients receiving a broad range of clinical doses (0.2 × 108/kg to 5 × 108/kg) of granulocyte transfusions observed in the first 12 to 18 hours after infusion a mean of 19.7% ± 17.4% DHR+ granulocytes (n = 15 subjects) for those without alloimmunization vs 0.95% ± 1.59% DHR+ granulocytes (n = 16) in those with alloimmunization.7 Most importantly, alloimmunization significantly increases the risks of graft rejection and failure if the patients undergo subsequent HSCT for definitive treatment of CGD.8,9 Although autologous cell infusions are advantageous in avoiding the development of anti-HLA immune responses, there is a potential for the development of immune responses against the novel proteins (p47phox or gp91phox), which may increase risks for future infusions, or even gene therapy or allogeneic HSCT. However, the same risks apply following gene therapy with autologous gene-corrected hematopoietic stem/progenitor cells and to date, there have not been any reports of immune responses in over 100 patients treated with gene therapy. Close monitoring of patients after receiving mRNA-electroporated cells will be important for future studies.
Granulocytes have a short half-life of 6 to 90 hours from egress from the marrow into the bloodstream and are prone to activation.18 Recent imaging studies that tracked the migration of neutrophils to the lungs, before homing to the bone marrow, suggest a much longer lifespan of several days.26 This raises potential concerns that transfected granulocytes may gravitate to the lungs following infusion and cause pulmonary complications. Indeed, we showed a higher percentage of human CD45+ cells in the lungs than in blood, liver, or spleen at 72 hours postinfusion into mice. To address this concern, we performed preclinical studies in NHPs (rhesus macaques) and included serial chest-imaging studies postinfusion that demonstrated no detectable effects on lungs. Nevertheless, gradual dose-escalation studies would be important for the eventual monitoring of infusion of mRNA-transfected cells into human patients. In addition to our in vitro data showing detectable protein expression in electroporated granulocytes out to 5 days, our data in rhesus monkeys also confirm persistence of protein expression in vivo in circulating granulocytes out to 5 days following transfusion, and confirm that multiple collections and infusions of mRNA-transfected cells are well tolerated.
A limitation of the study is the lack of an animal model to evaluate killing of microbes by electroporated phagocytes to verify their reconstituted function in vivo beyond demonstration of superoxide production with the DHR assay, and in vitro antifungal activity. We successfully completed performance validation runs in 5 donors (3 healthy volunteers and 2 CGD patients) showing consistent efficacy for the generation of Chemistry Manufacturing and Controls (CMC) information for US Food and Drug Administration (FDA) application.
The therapeutic mRNA electroporated into cells is nonintegrating and undergoes rapid degradation, which makes this approach of transient gene therapy relatively safe with respect to potential genotoxicity or extended action by the therapeutic mRNA.12 These include design features such as the use of modified nucleosides like pseudouridine.27 Although mRNA is generally degraded rapidly, the resulting protein may survive longer, conferring longer-term effects. The protein-encoding exogenous mRNAs were designed for optimal protein expression and manufactured to minimize immunogenicity. Consistent with the excellent cell viability, the NanoString data confirmed that there was no significant activation of the TLR-signaling pathway with delivery of the exogenous mRNA or inflammatory response genes. Of note, NADPH oxidase activity following p47phox or gp91phox mRNA correction was maintained out to 5 days (Figures 2E and 3C). The therapeutic effect of such transiently corrected granulocytes could be extended by repeated transfections and administration.
Chronic uncontrolled infections or viremia prior to HSCT are poor prognostic factors in multiple PIDs such as CD40L deficiency or SCID-X1.28,29 Our data showing efficient restoration of autologous PB immune cell function may bypass issues of alloimmunization and provide a short-term cellular therapy with functionally corrected autologous immune cells to control infections in patients with PIDs and stabilize patients as they await HSCT.
For original data, please contact sderavin@nih.gov.
Acknowledgments
The authors thank the National Institutes of Health veterinary and support staff for animal care. The authors thank their patients, their healthy volunteers, the Laboratory of Clinical Immunology and Microbiology nursing staff, the National Heart, Lung, and Blood Institute animal care staff, and the staff of the Dowling Apheresis Unit, Cell Processing Section, and Product Development Sections of the National Institutes of Health Clinical Center Department of Transfusion Medicine for their contributions to this study. The authors also thank MaxCyte Inc for their support and collaboration in the development of this project and transition to GMP-compliant manufacture of mRNA-corrected granulocytes for a clinical trial.
This work was supported in part by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases under projects Z01-AI-000988, Z01-AI-000644, and Z01-AI-001175-01.
Authorship
Contribution: S.S.D.R. and H.L.M. conceived the mRNA transfection of granulocytes correction concept and designed experiments; S.S.D.R., J.B., T.Q.L., M.A.Z., L.L., J.V.D., H.L., and S.L.H. performed the experiments or analyzed the data; N.T. assisted with sample preparation; R.J.M., A.B.C., and G.A.D. produced the mRNA; S.K., C.C., U.C., J.B., T.Q.L., A.C.B., M.E.M., and R.E.D. assisted with animal experiments; S.S.D.R. and J.B. helped write the manuscript with edits from the other authors; D.F.S., C.E.D., H.L.M., R.E.D., and J.F.T. provided support for the rhesus program; K.W., E.K., and D.F.S. provided advice for apheresis and transfusion product handling; D.B.K. assisted with CGD patient genetic diagnosis; and M.S.L., M.A.Z., and J.V.D. assisted with fungal studies.
Conflict-of-interest disclosure: L.L. is a full-time employee of MaxCyte Inc (Gaithersburg, MD). R.J.M., A.B.C., and G.A.D. are full-time employees of CELLSCRIPT, LLC (Madison, WI). The remaining authors declare no competing financial interests.
Correspondence: Suk See De Ravin, Laboratory of Clinical Immunology and Microbiology, National Institutes of Health, Building 10 CRC, Bethesda, MD 20892-1456; e-mail: sderavin@niaid.nih.gov; and Harry L. Malech, Laboratory of Clinical Immunology and Microbiology, National Institutes of Health, Building 10 CRC, Bethesda, MD 20892-1456; e-mail: hmalech@niaid.nih.gov.



