Key Points
IRA is an unrecognized fatal complication of allo-HSCT not reported before.
Incidence of IRA is 1% and IRA is fatal in 63% of cases.

Abstract
At our center, we observed a series of patients who developed transudative refractory ascites secondary to noncirrhotic, non-veno-occlusive disease (VOD)–related portal hypertension after allogeneic hematopoietic stem cell transplantation (allo-HSCT). Patients were considered to have idiopathic portal hypertension-related refractory ascites (IRA) if they developed ascites secondary to intrahepatic portal hypertension (serum ascites albumin gradient ≥1.1 g/dL or hepatic venous pressure gradient [HVPG] >5 mm Hg), but did not meet the clinical criteria for classical VOD/sinusoidal obstructive syndrome (SOS) and did not have any alternate etiology of portal hypertension. From our institutional database, we identified 40 patients who developed IRA after allo-HSCT between 2004 and 2018. The patients’ median age at the time of allo-HSCT was 54 years (range, 21-73 years). The median time to development of IRA after allo-HSCT was 80 days (range, 16-576 days). The median number of paracentesis was 3 (range, 1-11), and 15 (38%) patients had an intraperitoneal catheter placed for continued drainage of the rapidly accumulating ascites. Portal pressures were measured in 19 patients; 6 (15%) had moderate portal hypertension (HVPG 6-9 mm Hg), and 13 (33%) had severe portal hypertension (HVPG ≥ 10 mm Hg). Liver biopsy was performed in 24 patients. None of the patients met the criteria for classical VOD/SOS (clinical/histological) or cirrhosis (histological). The cumulative incidence of nonrelapse mortality was 63%, and the median survival duration after the development of the IRA was 7 months (range, 0.8-125.6 months). IRA is a poorly understood and often fatal complication of allo-HSCT.
Introduction
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is the potentially curative standard treatment of a variety of hematological malignancies. Conventional (myeloablative) allo-HSCT relies on the use of high-intensity conditioning chemotherapy to eradicate the malignant cells, whereas reduced-intensity conditioning relies more on the graft-versus-tumor effect. Regardless of the conditioning approach used, many recipients of allo-HSCT experience organ toxicity, which can be fatal. One significant contributor to organ toxicity is hepatic dysfunction, which occurs in approximately 18% to 83% of patients who have undergone allo-HSCT, depending on the intensity of the conditioning regimen.1-4
The rates of mortality from hepatic dysfunction are variable and depend on the etiology. Although veno-occlusive disease (VOD)/sinusoidal obstructive syndrome (SOS), acute graft versus host disease (GVHD), and drug-induced liver injury (DILI) occur early after HSCT, chronic GVHD, iron overload, and end-stage liver disease tend to occur in long-term survivors of HSCT. Depending on its etiology, liver injury can manifest in several ways.5,6 Acute liver insult can result in hepatocellular injury or cholestasis, leading to an increase in liver enzyme and bilirubin levels. Acute-on-chronic liver injury or chronic liver injury can lead to jaundice, hepatic microcirculatory dysfunction, an increase in hepatic vascular resistance, and development of portal hypertension. Portal hypertension and inappropriate renal retention of sodium are central to the pathogenesis of ascites formation. Ascites associated with portal hypertension can result from activation of the renin-angiotensin-aldosterone system or from an increase in hydrostatic pressure leading to transudation of fluid.7
The rate of occurrence of ascites after allo-HSCT has not been definitively determined. Most of the published cases of ascites after allo-HSCT occurred in patients who had VOD/SOS. Historically, the incidence of VOD/SOS is reported to be between 5% and 70% among bone marrow transplant recipients.8-11 This wide range is a result of variability in diagnostic criteria, patient ages, prior treatments, and the conditioning regimens used. More recent estimates place the incidence of VOD/SOS at around 14%12 ; this decrease is likely a result of the increased use of reduced-intensity conditioning regimens and improvements in our understanding and control of the risk factors for VOD/SOS. The incidence of ascites in patients with VOD/SOS is reported to be from 5% to 48%, depending on the severity of the VOD/SOS,10,13,14 with a higher incidence in patients with severe VOD/SOS. Isolated case reports of transudative ascites or serositis associated with chronic GVHD also exist.15
At our center, we observed a series of patients with hematological malignancies who developed post–allo-HSCT transudative refractory ascites secondary to portal hypertension. These patients had a high mortality rate, and none of them met the diagnostic criteria for VOD/SOS or had any systemic causes of portal hypertension that would explain the development of ascites. Most of them underwent a liver biopsy, and none of the biopsy specimens had the histological features of VOD/SOS, cirrhosis, or any infiltrative disease. In this retrospective study, we sought to determine the natural history, pathogenesis, and etiology of idiopathic portal hypertension-related ascites (IRA) developing after allo-HSCT.
Methods
Patient selection
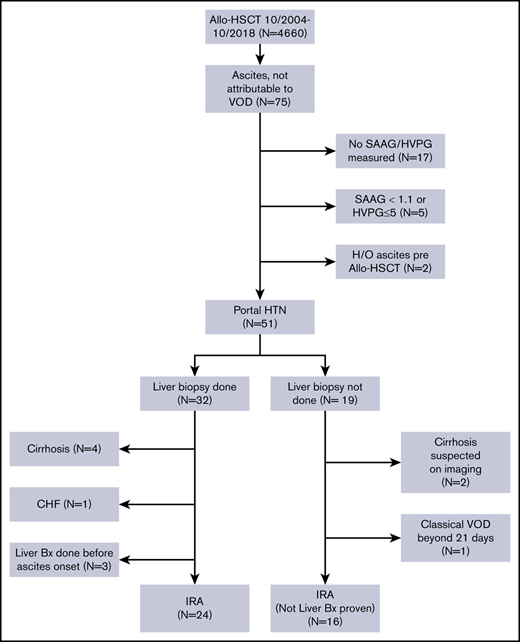
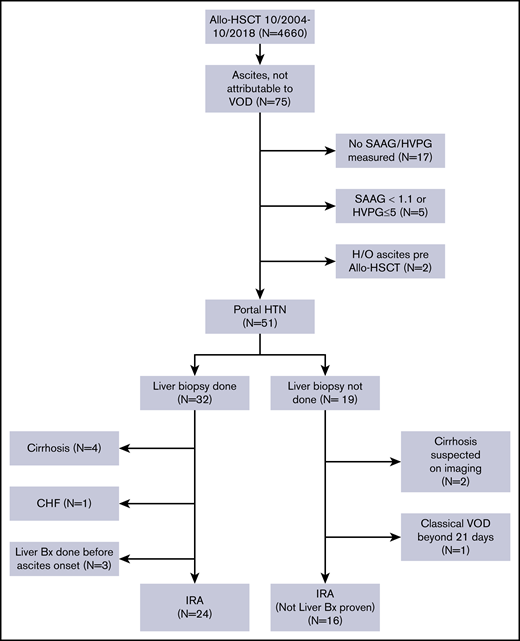
After obtaining approval from the Institutional Review Board of The University of Texas MD Anderson Cancer Center, we performed a retrospective review of the medical records of patients with hematological malignancies who underwent allo-HSCT at MD Anderson between October 2004 and October 2018. The requirement for informed consent was waived because of the retrospective nature of the study. We selected all patients aged 18 years or older who did not have VOD/SOS and who developed ascites (detected on clinical exam and confirmed with radiological tests) after allo-HSCT. A Consolidated Standards of Reporting Trials (CONSORT) diagram showing the inclusion and exclusion criteria for our study is shown in Figure 1.
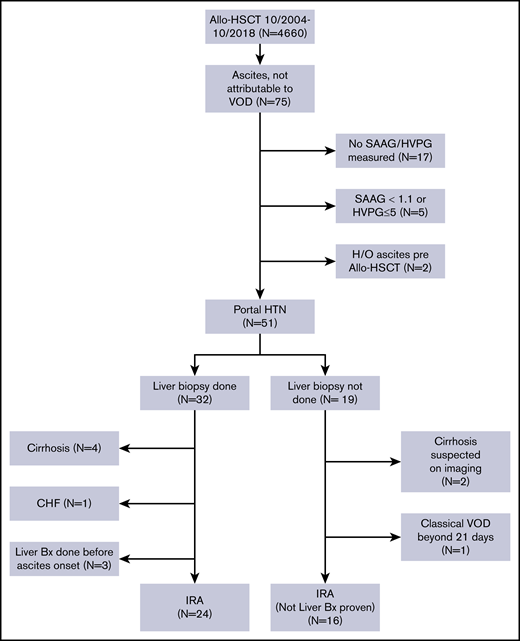
A total of 4660 patients aged 18 years and older underwent allo-HSCT at our institution during this time. Of them, 75 patients developed ascites not attributable to VOD/SOS. Patients whose ascitic fluid was not sent for diagnostic studies (n = 17), patients whose ascites was not related to portal hypertension (n = 5), and patients who had evidence of ascites on pre–allo-HSCT imaging studies (n = 2, patients with myelofibrosis [MF]) were excluded from the study. The remaining 51 patients all had portal hypertension-related ascites, which was defined as a hepatic venous pressure gradient (HVPG) >5 mm Hg or a serum ascites albumin gradient (SAAG) of at least 1.1 g/dL. Of these patients, 32 (63%) had a liver biopsy as part of the evaluation for elevated liver enzyme levels or to guide the management of ascites. Four patients who had cirrhosis on biopsy, 3 patients who underwent liver biopsy before the onset of ascites, and 1 patient who developed ascites in the setting of congestive heart failure were not considered to have IRA and were excluded from the study. Nineteen patients did not undergo a liver biopsy at any time during the duration of ascites. Two of them had imaging findings that were consistent with cirrhosis, and 1 patient had classical VOD/SOS beyond 21 days.16 These 3 patients were also excluded from the study. Thus, a total of 40 patients with IRA were included in the analysis.
Definitions
Patients were considered to have IRA if they developed ascites secondary to intrahepatic portal hypertension, but did not meet the clinical criteria for classical VOD/SOS, and if there was no alternate etiology for portal hypertension. We conducted a detailed retrospective chart evaluation to rule out other potential causes of portal hypertension, including congestive heart failure/constrictive pericarditis, alcoholic hepatitis, Budd-Chiari syndrome, and portal vein thrombosis.
Classical VOD/SOS was defined according to the Baltimore: a bilirubin level of at least 2 mg/dL and at least 2 of the following: hepatomegaly, ascites, or weight gain of more than 5% over baseline developing within 21 days after stem cell transplantation.17 For late-onset VOD/SOS, the European Society for Blood and Marrow Transplantation (EBMT) criteria were used (classical VOD/SOS beyond day 21, or histologically proven VOD/SOS, or 2 or more of the following: bilirubin level of at least 2 mg/dL, painful hepatomegaly, weight gain of more than 5%, or ascites, and hemodynamic or ultrasound evidence of VOD/SOS).16
Concurrent GVHD was considered new-onset GVHD requiring treatment and diagnosed within 1 month before the onset of ascites or worsening of existing GVHD requiring increased use of immunosuppressants.
Recurrent ascites was defined in our study population as the reappearance of ascites within 4 weeks of paracentesis and also being diuretic resistant.18
Liver function tests normal values
The upper limits of normal for baseline liver-function tests and ferritin that we used were 40 U/L for aspartate aminotransferase, 41 U/L for alanine aminotransferase, and 1.2 mg/dL for bilirubin.
Liver biopsy histology
Previously obtained liver biopsies had been processed routinely after formalin fixation. Two hematoxylin and eosin stained sections and a Masson trichrome stain were performed routinely, and reticulin staining was obtained when the histology suggested nodular regenerative hyperplasia. Liver biopsies were reviewed by a gastrointestinal/liver pathologist (S.C.A.). For the purposes of the current study, the presence or absence of the following histologic features was evaluated: fibrosis, nodular regenerative hyperplasia pattern of hepatic cords, aberrant periportal vessels, narrowed and/or inapparent portal venules, sinusoidal dilatation, and GVHD. Other findings (eg, steatosis) were recorded as appropriate.
Statistical analyses
All data are expressed as frequencies, percentages, medians, and ranges, as appropriate. Ascites overall survival (OS) was calculated from the date of onset of ascites to the date of death from any cause. Transplant OS was calculated from the date of allo-HSCT to the date of death from any cause. Patients who were alive on the last date of follow-up were censored in the analysis. Kaplan-Meier method was used to estimate median survival durations and to generate the survival curves. All statistical analyses were performed with Stata software, release 15 (Stata Corporation, College Station, TX,) and the survival curves were generated from XLSTAT 2019.1.
Results
Patient characteristics
We identified 40 patients (0.9%) out of the 4660 patients who underwent allo-HSCT at our center and met the criteria for IRA. The baseline characteristics of these patients are summarized in Table 1. The patients’ median age was 54 years (range, 21-73 years). Among the 40 included patients, 16 (40%) had acute myeloid leukemia or myelodysplastic syndrome; 12 (30%) had non-Hodgkin lymphoma, Hodgkin lymphoma, or chronic lymphocytic leukemia; 11 (28%) had acute lymphoblastic leukemia; and 1 (2%) had MF.
Conditioning regimen
Twenty-five (63%) patients had a myeloablative conditioning regimen, and 15 (37%) patients had a reduced-intensity or nonmyeloablative conditioning regimen. The most common conditioning regimen used was fludarabine in combination with melphalan, in 14 (35%) patients. Nineteen (48%) patients received a busulfan-based conditioning regimen, 2 (5%) patients underwent total body irradiation, and 1 (3%) patient received gemtuzumab as a part of the conditioning regimen.
Pre–allo-HSCT liver-function status
All patients with IRA had undergone testing for hepatitis B surface antibodies, hepatitis B surface antigens, hepatitis B core antibodies, and hepatitis C antibodies before allo-HSCT; 2 patients had positive hepatitis B core antibody and 1 had positive hepatitis B surface antigen and detectable hepatitis B DNA (38 IU/mL). Hepatitis C antibody was positive in 1 patient, but hepatitis C RNA was undetectable (Table 2). Liver imaging studies were conducted in 29 patients within 3 months before allo-HSCT; 1 patient had hepatomegaly (Myelofibrosis). Ultrasonography in 1 patient showed increased echogenicity of the liver, which was suggestive of fatty infiltration. One patient had been treated for a primitive neuroendocrine tumor of the liver 3 years before allo-HSCT.
The median bilirubin level in the patient cohort was 0.6 mg/dL, and no patients had a bilirubin level of more than 1.5 times the upper limit of normal. Three patients had aspartate aminotransferase levels of more than 2.5 times the upper limit of normal, and 4 patients had alanine aminotransferase levels of more than 2.5 times the upper limit of normal before allo-HSCT (Table 2). One patient had a liver biopsy 3 months before allo-HSCT; the histology was consistent with fatty liver disease.
Follow-up and ascites development
The median time of onset of ascites from the date of allo-HSCT was 80 days (range, 16-576 days). SAAG was measured in 37 patients, in whom the median SAAG was 1.7 g/dL (range, 1.1-2.8 g/dL). The median ascitic fluid protein level was 2.0 g/dL (range, 0.9-3.7 g/dL). The median serum bilirubin level at the time of onset of ascites was 0.8 mg/dL (range, 0.4-3.2 mg/dL). All patients were refractory to the treatment of sodium restriction and diuretic therapy. The median number of paracentesis procedures performed was 3 (range, 1-11), and an intraperitoneal catheter for ascitic fluid drainage was placed in 15 (38%) patients. Two patients also underwent placement of a transjugular intrahepatic portosystemic shunt (TIPS) to relieve the refractory ascites and portal hypertension. None of the patients received defibrotide. Liver biopsy was performed in 24 (67%) patients at a median of 29 days after the onset of ascites (range, 0-853 days). None of those patients had the histological features of VOD/SOS: cirrhosis (details of histology are mentioned in "Histologic findings"). The median HVPG among the 19 (48%) patients in whom it was measured was 10.5 mm Hg (range, 7-17 mm Hg); 6 (15%) patients had moderate portal hypertension (HVPG 6-9 mm Hg), and 13 (33%) patients had severe portal hypertension (HVPG ≥ 10 mm Hg; Table 3). The median number of paracentesis in the HVPG 6 to 9 mm Hg group was 2 (range, 1-6), and in the 10 mm Hg or more group was 3 (range, 1-8). Intraperitoneal catheter for ascitic fluid drainage was placed in 1 patient in the 6 to 9 mm Hg HVPG group, and in 5 patients (38%) in the 10 mm Hg or more group.
Esophageal or splenic varices were detected on either esophagogastroduodenoscopy or imaging in 7 (18%) patients, and 17 (43%) patients had an enlarged spleen on imaging. Doppler ultrasonography was performed in 36 (90%) patients. In all cases, it demonstrated normal flow to the liver, and no reversal of flow in the portal and hepatic veins was identified. Transthoracic echocardiograms were performed in 23 (58%) patients; no right or left ventricular dysfunction was identified. Cytologic examinations revealed that the ascitic fluid did not contain malignant cells in any of the patients. Ten (25%) patients had concurrent pleural effusion diagnosed at the same time as ascites, and 4 of them required thoracentesis. The pleural fluid was exudative in 1 patient and transudative in 3. Three (8%) patients had concurrent pericardial effusion, which resolved on its own in 2 of them; 1 patient underwent pericardiocentesis and was treated with colchicine. The pericardial fluid was exudative in that patient.
Histologic findings
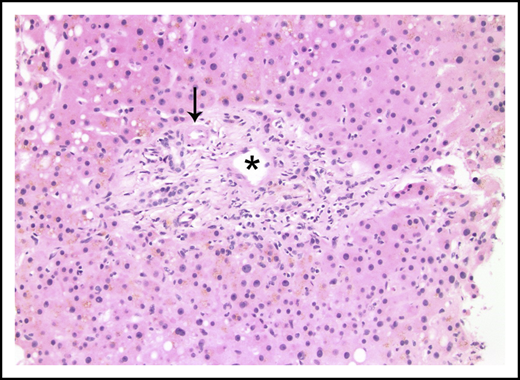
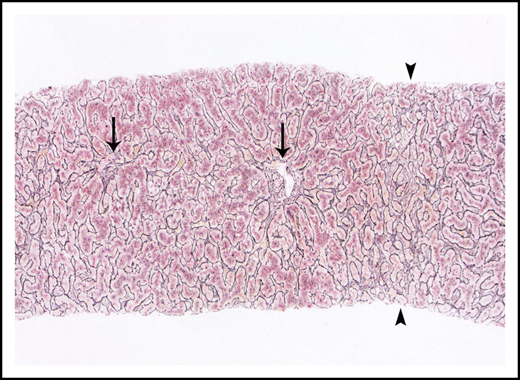
Liver biopsies were performed in 24 patients. Fibrosis was variable, but no patient had cirrhosis. Ten (42%) had no fibrosis, 6 (25%) had mild perisinusoidal fibrosis only, 5 (21%) had periportal fibrosis (4 of these additionally demonstrated perisinusoidal fibrosis), 2 (8%) had bridging fibrous septae, and 1 (4%) had portal fibrosis only. Portal and parenchymal changes classically associated with IRA included narrowed or inapparent portal venules (Figure 2) in 10 (42%) biopsies, nodular regenerative hyperplasia (Figure 3) in 8 (33%), and aberrant periportal vessels (Figure 4) in 5 (21%). Fourteen (58%) biopsies showed sinusoidal dilatation. Other abnormalities included fatty liver disease in 3 (13%) patients; 1 of these had moderate macrovesicular steatosis, 1 had moderate macrovesicular steatosis and mild (grade 1 of 3) steatohepatitis, and the third had moderate to severe (grade 2-3) steatohepatitis with central-portal bridging fibrosis. The remaining 21 (87%) patients had no or minimal (<5%) steatosis. One (4%) additional biopsy contained patchy extramedullary hematopoiesis within sinusoids. Features of concomitant acute GVHD are presented in "Concurrent GVHD with ascites." None of the biopsies had established ductopenia (ie, chronic GVHD). Two biopsies had histological features of DILI (1 had cholestasis and the other had cholestasis and zone 3 [centrilobular] hepatocyte dropout/necrosis). Three patients also had their liver examined at autopsy, and none of them had histological features of VOD/SOS or cirrhosis.
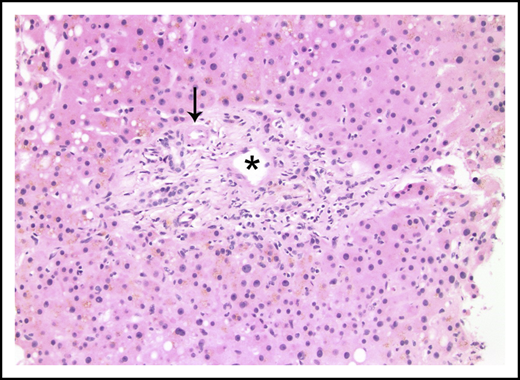
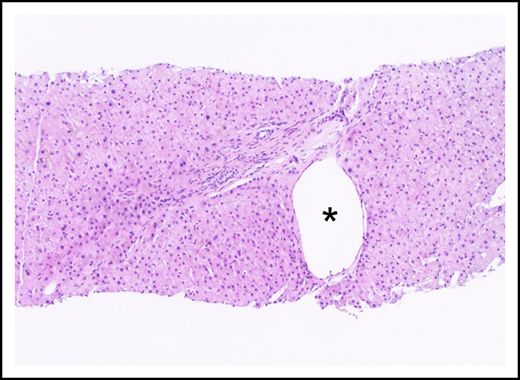
Portal tract containing a narrowed portal venule (asterisk), which is barely larger than the nearby arteriole (arrow; hematoxylin and eosin stain).
Portal tract containing a narrowed portal venule (asterisk), which is barely larger than the nearby arteriole (arrow; hematoxylin and eosin stain).
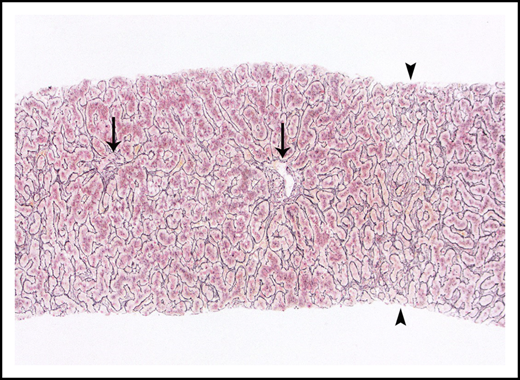
Nodular regenerative hyperplasia pattern by reticulin stain, which serves to outline hepatic cords. Hepatic cords are thickened around the portal tracts (arrows) and are atrophic in zone 3 (arrowheads).
Nodular regenerative hyperplasia pattern by reticulin stain, which serves to outline hepatic cords. Hepatic cords are thickened around the portal tracts (arrows) and are atrophic in zone 3 (arrowheads).
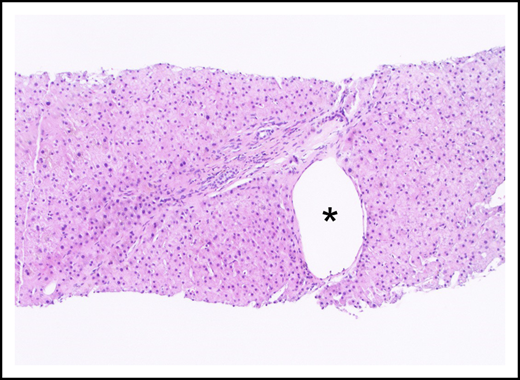
An aberrant periportal venule (asterisk) extends beyond the confines of the portal tract (hematoxylin and eosin stain).
An aberrant periportal venule (asterisk) extends beyond the confines of the portal tract (hematoxylin and eosin stain).
Concurrent GVHD with ascites
Ten (25%) of the 40 patients had concurrent acute GVHD of the skin and/or gastrointestinal tract at the time of diagnosis of ascites. Of the 24 patients undergoing a liver biopsy, histologic findings supported a clinical suspicion of acute liver GVHD in 4 (17%). Three of these biopsies showed mild interlobular duct epithelial injury, focal lymphocytic cholangitis, and mild mixed portal inflammation; the fourth showed more overt duct injury with occasional (but <50%) ductopenic portal tracts. None of the patients with IRA had diagnostic or distinctive features of concurrent chronic GVHD at the time of diagnosis of ascites.
Outcomes
Ascites resolved in 16 (40%) patients at a median duration of 3.7 months (range, 0.5-32.6 months) from diagnosis; 3 of the 6 patients in the HVPG 6 to 9 mm Hg group had resolution of ascites at a median of 70 days (range, 30-181 days); 4 of the 13 patients in patients in the HVPG 10 mm Hg or higher group had resolution of ascites at a median of 393 days (range, 270-979 days). In 24 (60%) patients, the ascites did not resolve; 23 of these patients had died at the time of the last follow-up. The 1 patient who was alive at last follow-up, the ascites resolved after TIPS placement but reappeared after 44 months; this patient’s liver imaging is now consistent with cirrhosis.
Nonrelapse mortality and OS
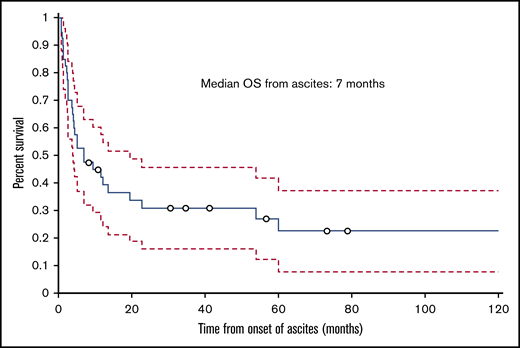
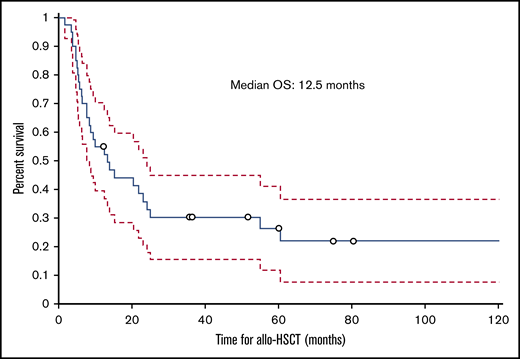
The median follow-up time from the time of transplant was 12.5 months (range, 1.8-129.2 months); 30 (75%) patients died during this time. The cumulative incidence of nonrelapse mortality was 63% for all patients, 50% in the HVPG 6 to 9 mm Hg group, and 77% in the HVPG 10 mm Hg or more group. Ten patients died of infection, 6 of complications of ascites or liver failure, 3 of GVHD, 2 of multiple organ failure, and 2 of other causes (eg, secondary malignancy, cardiogenic shock). The cause of death was not known for 2 patients. Of the 6 patients who died of ascites or liver failure, 3 patients died of liver failure, 1 of whom had liver failure as a complication of TIPS. Among the 3 patients who died of complications of ascites, 2 died of hemoperitoneum or hemorrhage as a complication of the biopsy of the liver and peritoneum to determine the etiology of ascites, and 1 died of peritonitis. The median ascites OS duration for all patients with IRA was 7 months (range, 0.8-125.6 months; Figure 5), and the median transplant OS duration was 12.5 months (1.8-129.2 months; Figure 6).
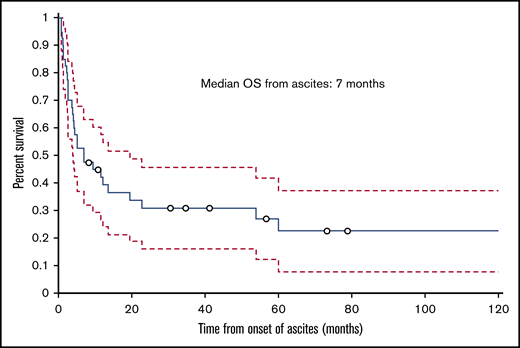
Kaplan-Meier curve showing OS of patients with IRA from the date of onset of ascites.
Kaplan-Meier curve showing OS of patients with IRA from the date of onset of ascites.
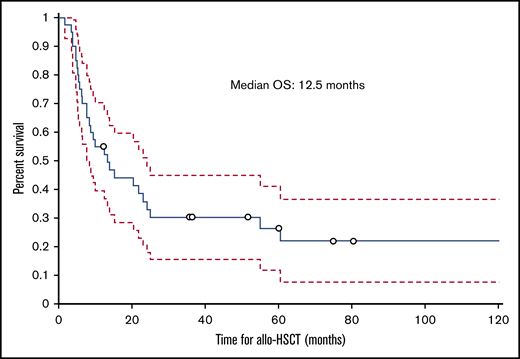
Kaplan-Meier curve showing OS of patients with IRA from the date of allogeneic hematopoietic stem-cell transplantation.
Kaplan-Meier curve showing OS of patients with IRA from the date of allogeneic hematopoietic stem-cell transplantation.
Discussion
Ascites secondary to portal hypertension after allo-HSCT is not well documented in the literature. In the early period (≤21 days) after allo-HSCT, VOD/SOS is the most common reported cause, but little is known about the etiologies of ascites that develops after 21 days. A few case reports and case series discuss late-onset VOD/SOS as a cause of ascites,19-21 but not much is known about other etiologies. A few case reports on other etiologies of ascites after allo-HSCT exist,15,22-24 but most of them do not report a detailed workup for determining the etiology of ascites. Here, we report the clinical characteristics and outcomes of patients with hematological malignancies who developed the IRA after allo-HSCT.
The etiology of IRA remains unknown, and it needs to be differentiated from some of the other known causes of post–allo-HSCT portal hypertension-related ascites.
Why is it not VOD/SOS?
Two sets of criteria are commonly used to diagnose VOD/SOS: the modified Seattle Criteria9 and the Baltimore criteria.17 However, the sensitivity and specificity of these criteria are not well documented. Under both these criteria, patients need to develop symptoms of VOD/SOS (ascites, weight gain, hepatomegaly, bilirubin ≥2 mg/dL) within 20 or 21 days of allo-HSCT. In our patient cohort, only 2 patients had developed ascites by 21 days after allo-HSCT, and these patients did not have a bilirubin level of 2 mg/dL or higher, or hepatomegaly on imaging. Thus, the diagnostic criteria for early-onset VOD/SOS may not be useful in determining the etiology of ascites in our patients with IRA.
Late-onset VOD/SOS has been reported in a few case series and studies.19-21 Pai et al19 reported 8 patients with late-onset VOD/SOS, one of the largest series focusing primarily on late-onset VOD/SOS. VOD/SOS was diagnosed in those patients at a median of 52 days (range, 33-77 days). All 8 of them had clinical features consistent with VOD/SOS, and 7 also had histological confirmation of the diagnosis by liver biopsy. Recently, the EBMT came up with criteria for the diagnosis of late-onset VOD/SOS.16,21,25 Anicteric VOD/SOS in adults is rare, and the majority of the patients in both the studies by Lee et al25 and Carreras et al,21 (87% and 100%, respectively) had bilirubin levels of 2 mg/dL or higher. Thirteen percent of patients who did not have elevated bilirubin in the study by Lee et al25 also did not have histological confirmation of VOD/SOS. In contrast, in our study, the majority of our patients did not have hyperbilirubinemia, and the 5 patients who had bilirubin levels of 2 mg/dL or higher had a liver biopsy that was not consistent with VOD/SOS. Normal bilirubin level/anicteric is often associated with late-onset VOD/SOS,26 and one might wonder whether these patients actually have IRA and not late-onset VOD/SOS, especially if there is no histological confirmation. Carreras et al,21 reported 19 patients with VOD/SOS in the busulfan/melphalan arm, including 14 patients with late-onset VOD/SOS. Five of the 19 patients (26%) died; most of the patients had a nonfatal course (11/19, 52% of whom had complete resolution of the symptoms of VOD/SOS in 17 days). On the contrary, our patients with IRA had a much aggressive course. Cumulative incidence of NRM was 63%, and in the HVPG 10 mm Hg or higher group, it was 77%. Ascites did not resolve in 60% of the patients, and in the patients in whom it did resolve (40%), it resolved at a median of 3.7 months. The median time to onset of ascites in our patients with IRA was also somewhat longer than in previous reports, at 80 days, and many of our patients (N = 19, 48%) with IRA met the EBMT criteria for late-onset VOD/SOS (ascites, weight gain, and abnormal HVPG). However, all of those patients underwent a liver biopsy, and an autopsy was performed in 1 patient; none of them had histological features of VOD/SOS. The current EBMT criteria for late-onset VOD/SOS can overestimate its diagnosis, as any patient with abnormal HVPG (hemodynamic evidence of VOD/SOS), ascites, and weight gain after HSCT would be classified as late-onset VOD/SOS. Histological confirmation of VOD/SOS might not be necessary if the cases have classical features of VOD/SOS, but it is important to rule out alternate etiologies if the diagnosis of VOD/SOS is questionable, especially if it is anicteric and occurs late after HSCT.
The histopathology of VOD/SOS and other syndromes that result in venous outflow obstruction is well-described.27,28 Liver biopsies with VOD/SOS show a characteristic pattern of injury that is predominantly centrilobular in distribution. VOD/SOS results in loss of endothelial cells and sinusoidal lining cells, accumulation of fibrin and entrapped red blood cells within the central veins, and variable pressure necrosis of centrilobular hepatocytes. None of these was present in liver biopsies from patients with IRA. It is true that sinusoidal dilatation and (more chronically) fibrosis are histologic features of VOD/SOS, as well as features of IRA, but the characteristics of each feature are different in VOD/SOS and in IRA. The sinusoidal dilatation in VOD/SOS is greatest around the central veins and is accompanied by red blood cell congestion and extravasation of erythrocytes through the damaged sinusoidal lining, into the hepatic plates. In contrast, sinusoidal dilatation in our cases was not secondary to venous outflow obstruction, and therefore did not show red blood cell congestion or extravasation of red blood cells through the sinusoidal lining. Likewise, fibrosis in VOD/SOS mirrors the centrilobular injury of that process and can result in a central vein-to-central vein or central vein-to-portal fibrous bridges. The fibrosis in our biopsies, when present, was portal-based and was accompanied by variable portocentric abnormalities including narrowed portal venules (present in 42% of our biopsies), nodular regenerative hyperplasia (33%), and aberrant periportal vessels (21%). Notably, although fibrosis, sinusoidal dilatation, narrowing of portal venules, nodular regenerative hyperplasia, and aberrant periportal vessels are common findings in IRA and in idiopathic noncirrhotic portal hypertension,29 each of the features is only variably present and none is entirely specific. In contrast, the histologic injury of VOD/SOS forms a highly characteristic pattern that is absent in our biopsies from patients with IRA.
Why is it not serositis due to chronic GVHD?
Serositis is an uncommon manifestation of chronic GVHD, with an incidence of 0.9% to 1.8% after allo-HSCT.23,30,31 The fluid accumulated in serositis is mostly transudative. Serositis most commonly presents as polyserositis, with a combination of pleural effusion, pericardial effusion, and ascites, but a few cases of isolated effusion have also been reported.15 In a study of the causes of pleural effusion after allo-HSCT in 71 patients with hematological malignancies, Modi et al32 found that 8 patients’ pleural effusion was caused by serositis/chronic GVHD, and 2 had concurrent ascites with the pleural effusion. Most of these patients had extensive chronic GVHD before the onset of pleural effusion, with a median time to onset of pleural effusion from allo-HSCT of 400 days. Bagal et al15 also reported 2 patients who developed ascites after allo-HSCT as a manifestation of chronic GVHD; however, these patients did not have concurrent pleural or pericardial effusion. At the time of diagnosis of IRA, none of our patients had diagnostic or distinctive features of concurrent chronic GVHD. In addition, none of our 24 patients who underwent liver biopsy had histological features of chronic GVHD. We did not have a liver biopsy for 16 of our patients, and ascites could have developed as a result of chronic liver GVHD in these patients; however, chronic liver GVHD/serositis is rarely the only manifestation of GVHD. Ten of our patients had pleural effusion detected at the same time as ascites. Four of these patients required thoracentesis, and the fluid was exudative in 1 and was transudative in the other 3. All 3 patients with transudative pleural effusion had hypoalbuminemia, which could have led to the development of pleural effusion in them.
Is IRA caused by drug-induced liver injury?
Because of polypharmacy, it is often challenging to determine which drug is responsible for DILI; we often depend on the proximity of the drug known to cause liver injury. Nonetheless, some drugs are known to cause DILI more often than others. Conditioning regimens containing cyclophosphamide, busulfan, and melphalan, as well as concomitant myelosuppressive medications such as methotrexate and sirolimus, are known to increase the risk for VOD/SOS.33 Other drugs, including antifungals (eg, amphotericin, azoles) and antibacterials (eg, trimethoprim-sulfamethoxazole), can cause mild to moderate DILI, but are rarely associated with severe liver injury.33 None of these drugs is associated with the development of portal hypertension. Of our patients with IRA, 36 (90%) received conditioning chemotherapy containing cyclophosphamide, busulfan, or melphalan. Twenty-five (63%) also received myeloablative conditioning. Of the 24 patients who underwent a liver biopsy, 2 patients had features of DILI on histopathologic examination. In these cases, DILI may have contributed to the development of IRA.
Is it related to the underlying hematological malignancy?
Portal hypertension in patients with hematological malignancies has not been well studied. Although it has been described in patients with MF in a few case series,34 few such descriptions are available for patients with lymphoproliferative diseases. The pathogenesis of portal hypertension in MF is unknown, but it likely develops because of increased intrahepatic resistance from extramedullary hematopoiesis and infiltration of the liver with myeloid cells. The increased blood flow through the portal vein as a result of the enlarged spleen in these patients can also contribute to portal hypertension. Abu-Hilal and Tawaker34 reported 13 patients with MF who developed portal hypertension at an interval of 1 to 11 years after the diagnosis of MF. Three of the 13 patients also had ascites. Dubois et al.35 performed a liver biopsy in 20 patients with myeloproliferative disease and 47 patients with lymphoproliferative disease; approximately 50% of these patients had evidence of portal hypertension, which the authors defined as HVPG of 6 mm Hg or higher. Hepatic infiltration by malignant cells was found in about 45% of patients in both the myeloproliferative disease and lymphoproliferative disease groups. Portal venous blood flow was also found to be significantly higher in both groups than in healthy volunteers.
Of the 51 patients in our study who had ascites related to portal hypertension, only 1 had MF. Two other patients with MF, who developed non-VOD/SOS–associated ascites after allo-HSCT, had a history of ascites before allo-HSCT and were excluded from our study. The vast majority (97%) of our patients with IRA did not undergo liver biopsy before allo-HSCT, and their pre–allo-HSCT liver imaging was not suggestive of malignant infiltration of the liver. Only 1 patient had a pre–allo-HSCT liver biopsy, performed 3 months before allo-HSCT; in this patient, the histology was consistent with fatty liver disease.
IRA is a rare, unrecognized vascular liver disease of unknown etiology, and ours is the first study to report 40 patients who developed IRA after allo-HSCT. Although cases of idiopathic noncirrhotic portal hypertension have been reported in non–allo-HSCT patients, and these patients can share the same histological features as IRA (fibrosis, sinusoidal dilatation, narrowing of portal venules, nodular regenerative hyperplasia, and aberrant periportal vessels), most of those patients have a good prognosis and have similar survival to that of the general population.36 The 10-year liver transplant-free survival can be close to 80% in them.37 Idiopathic portal hypertension and extrahepatic portal venous obstruction (EHPVO) are the 2 major causes of idiopathic noncirrhotic portal hypertension.38 Although idiopathic portal hypertension is primarily a disease of young to middle age and EHPVO is a disease of childhood, the median age of our patients was around 50 years. Also, the HVPG is normal in EHPVO and slightly elevated in idiopathic portal hypertension,38 whereas HVPG was significantly elevated in our patients. Ascites in idiopathic noncirrhotic portal hypertension patients is generally transient and very often resolves with controlling the trigger and with the use of diuretics.39,40 In contrast, our patients with IRA had a much worse prognosis and the NRM was 63% in all patients and 50% and 77% in the 6 to 9 mm Hg and 10 mm Hg or more HVPG groups, respectively. Also, ascites was refractory in nature in our patients and did not resolve in majority of our patients (60%), and one-third of our patients needed an intraperitoneal catheter for drainage of the rapidly accumulating recurrent ascites.
The etiopathogenesis of IRA is unknown and can be speculated based on the risk factors evaluated, liver histology and clinical course of our patients. Sixty-three percent of our patients with IRA received myeloablative conditioning regimen and 48% of patients received a busulfan-based conditioning regimen. The occurrence of IRA was not limited to any particular disease type (acute myeloid leukemia/myelodysplastic syndrome, acute lymphoblastic leukemia, non-Hodgkin lymphoma, MF), disease status at HSCT, prior HSCT, cell source, and donor type. We also looked at whether the pre–allo-HSCT liver function status affected the occurrence of IRA, but most of our patients with IRA (>90%) had normal liver function pre–allo-HSCT, preventing us from drawing any meaningful conclusion. The median HVPG was 10.5 mm Hg for our patients, and an elevated HVPG suggests an intrahepatic cause of portal hypertension. We speculate that the IRA is a result of hepatocellular injury resulting from conditioning chemotherapies, hepatotoxic chemotherapies, cytokine release from damaged tissues, and alloreactivity. Another possibility is that these patients might have had infiltration of the liver by malignant cells pre HSCT, which could have predisposed them to develop IRA, especially after myeloablative conditioning. We treated patients with IRA with sodium restriction, diuretic therapy, frequent paracentesis, intraperitoneal catheter placement, and transjugular intrahepatic portosystemic shunt. However, these therapies and interventions did not provide long-term control of symptoms, and most of the patients had a poor prognosis. Most of these cases are from the predefibrotide era, and the use of defibrotide to treat these patients remains speculative. IRA is a diagnosis of exclusion, and we propose the following criteria to diagnose IRA in recipients of allo-HSCT: (1) new-onset portal hypertension (HVPG >5 mm Hg or a SAAG ≥1.1 g/dL) related to ascites after allo-HSCT; onset of ascites in most cases after 21 days of allo-HSCT; (2) absence of VOD/SOS, cirrhosis, and other causes of chronic liver disease; and (3) patent hepatic and portal veins on imaging.
Our study has some limitations, including the inherent limitations of a retrospective study and a heterogeneous patient population. We recognize that with current diagnostic criteria of late-onset VOD/SOS,16 there is some gray zone between late-onset VOD/SOS and IRA, which is not easy to clarify. Both of them have ascites resulting from portal hypertension, and on the basis of clinical signs/symptoms and laboratory abnormalities, it is not possible to definitively say whether it is late-onset VOD/SOS or IRA. We also understand that our study is limited by a small sample size. However, the key differences between the 2 are the time to onset of symptoms, liver histology, and fatal prognosis of the patients with IRA. Ascites in IRA occurs much later in comparison with late-onset VOD/SOS.19-21 The median time to onset of ascites was 80 days (range, 16-576 days) for patients with IRA, with a few cases occurring even after 500 days of allo-HSCT. In contrast, most of the cases of late-onset VOD/SOS reported in literature19-21 have a much earlier time to onset of symptoms. Also, the liver histology in patients with VOD/SOS shows a characteristic pattern of centrilobular injury that is absent in patients with IRA. The fibrosis in VOD/SOS results in a central vein-to-central vein or central vein-to-portal fibrous bridges, and fibrosis, when present in IRA, is portal based. Furthermore, in VOD/SOS, the sinusoidal dilatation is greatest around the central veins and is accompanied by red blood cell congestion. In contrast, in patients with IRA, there is no venous outflow obstruction, and hence no erythrocyte congestion or extravasation occurs through the sinusoidal lining. We also understand that not all the patients with IRA in our cohort underwent liver biopsy. Thus, we could have missed some possible additional etiologies of portal hypertension, as not every patient was examined for alternate etiologies to the same extent. However, most of these alternative etiologies have very low incidence.41 Also, anicteric VOD/SOS in adults is rare.26 and all the patients who did not have a liver biopsy had normal bilirubin and lacked the ultrasound evidence of VOD/SOS, making the diagnosis of VOD/SOS unlikely. Uncommonly, the histological changes of early-onset VOD/SOS can be missed in a small sample of liver biopsy because of the patchy nature of the disease. Nonetheless, 3 of our patients had liver histology examined at autopsy, and none of them had features of VOD/SOS.
In conclusion, we report the occurrence of refractory ascites resulting from portal hypertension of unknown etiology in 40 patients after allo-HSCT. Furthermore, we describe the natural history of this entity. Further investigation is necessary to establish the pathophysiology, etiology, and treatment of these patients who develop this fatal complication after allo-HSCT.
E-mail data sharing requests to the corresponding author, Uday R. Popat (upopat@mdanderson.org).
Acknowledgment
The authors acknowledge the patients at MD Anderson, without whom this research would not have been possible.
Authorship
Contribution: A.V. collected data, performed statistical analysis, and wrote the paper; S.C.A. interpreted the liver histology and wrote a section of the article; R.S.M., M.H., S.A.S., Q.B., B.O., C.M.H., Y.N., P.K., A.M.A., S.A., D.M., I.F.K., S.O.C., M.H.Q., K.R., P.A., B.S.A., E.J.S., and R.E.C. edited the paper; N.Y.S. performed statistical analysis; M.R., J.C., and G.R. collected the data; U.R.P. designed the study, interpreted results, performed statistical analysis, and edited the paper; and all authors reviewed and approved the final version of the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Uday R. Popat, Department of Stem Cell Transplantation and Cellular Therapy, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Unit 0423, Houston, TX 77030; e-mail: upopat@mdanderson.org.