Key Points
EPO plays an important role in FGF23 signaling in hereditary hemolytic anemias.
There is a clear correlation between EPO and ERFE in patients with a variety of congenital hemolytic anemias other than hemoglobinopathies.

Abstract
Recently, erythropoietin (EPO) was identified as regulator of fibroblast growth factor 23 (FGF23). Proteolytic cleavage of biologically active intact FGF23 (iFGF23) results in the formation of C-terminal fragments (cFGF23). An increase in cFGF23 relative to iFGF23 suppresses FGF receptor signaling by competitive inhibition. EPO lowers the i:cFGF23 ratio, thereby overcoming iFGF23-mediated suppression of erythropoiesis. We investigated EPO-FGF23 signaling and levels of erythroferrone (ERFE) in 90 patients with hereditary hemolytic anemia (www.trialregister.nl [NL5189]). We show, for the first time, the importance of EPO-FGF23 signaling in hereditary hemolytic anemia: there was a clear correlation between total FGF23 and EPO levels (r = +0.64; 95% confidence interval [CI], 0.09-0.89), which persisted after adjustment for iron load, inflammation, and kidney function. There was no correlation between iFGF23 and EPO. Data are consistent with a low i:cFGF23 ratio. Therefore, as expected, we report a correlation between EPO and ERFE in a diverse set of hereditary hemolytic anemias (r = +0.47; 95% CI, 0.14-0.69). There was no association between ERFE and total FGF23 or iFGF23, which suggests that ERFE does not contribute to the connection between FGF23 and EPO. These findings open a new area of research and might provide potentially new druggable targets with the opportunity to ameliorate ineffective erythropoiesis and the development of disease complications in hereditary hemolytic anemias.
Introduction
Systemic erythropoietin (EPO) production plays a critical role in maintaining erythropoietic homeostasis.1 The amount of circulating EPO is elevated in most forms of hereditary anemia, although the amount is often still relatively low for the degree of anemia. Insight into components of the signaling cascade downstream of EPO might lead to a greater understanding of EPO responsiveness in individual patients. Recently, EPO was identified as an important regulator of production and cleavage of fibroblast growth factor 23 (FGF23).2-7 Because erythroid progenitor cells express high levels of FGF23 and carry the FGF receptor, it is likely that they are involved in the FGF23 metabolic pathway.
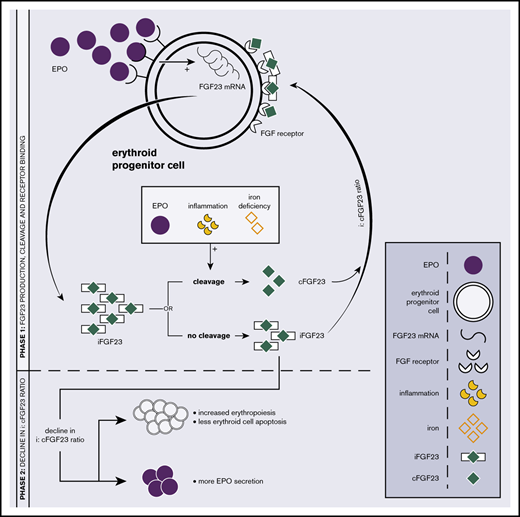
FGF23 was originally identified as a bone-derived hormone, a key player in phosphate and vitamin D metabolism, and a regulator of bone mineralization. It was shown that bone marrow, specifically the early erythroid lineage, contributes significantly to total circulating FGF23.2,5,8 Erythroid progenitor cells express FGF23 messenger RNA (mRNA) under physiologic conditions, and significant increases are observed in response to EPO.2,5,6 FGF23 is formed as intact biologically active protein (iFGF23). Proteolytic cleavage results in the formation of an assumed inactive C-terminal tail FGF23 (cFGF23). By competitive inhibition, an increase in cFGF23 relative to iFGF23 leads to suppression of FGF receptor signaling9,10 (Figure 1A; phase 1).
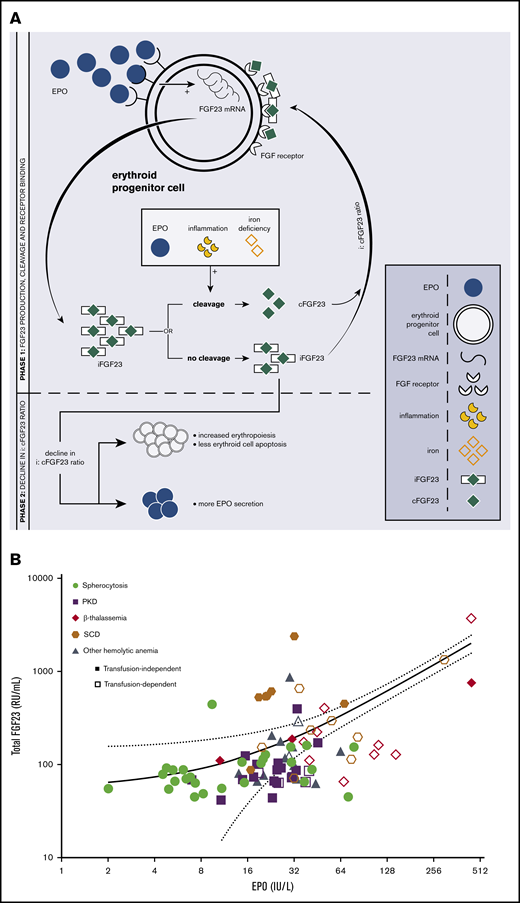
The relationship between EPO and FGF23 metabolism. (A) Schematic overview of the EPO-FGF23 signaling pathway in the erythroid lineage in bone marrow. Phase 1 shows FGF23 production, the secretory process, and FGF receptor binding; phase 2 summarizes the effects of a decrease in the i:cFGF23 ratio. (B) Correlation between C-terminal FGF23 and EPO. Individual patient data are represented as disease and transfusion requirement. Correlation between total FGF23 and EPO in patients with various hemolytic anemias: r = +0.64; 95% CI, 0.09-0.89. The solid line represents the linear regression line; the dashed lines are the 95% CI bands. PKD, pyruvate kinase deficiency; SCD, sickle cell disease.
The relationship between EPO and FGF23 metabolism. (A) Schematic overview of the EPO-FGF23 signaling pathway in the erythroid lineage in bone marrow. Phase 1 shows FGF23 production, the secretory process, and FGF receptor binding; phase 2 summarizes the effects of a decrease in the i:cFGF23 ratio. (B) Correlation between C-terminal FGF23 and EPO. Individual patient data are represented as disease and transfusion requirement. Correlation between total FGF23 and EPO in patients with various hemolytic anemias: r = +0.64; 95% CI, 0.09-0.89. The solid line represents the linear regression line; the dashed lines are the 95% CI bands. PKD, pyruvate kinase deficiency; SCD, sickle cell disease.
FGF23 knockouts or animals treated with iFGF23-blocking peptides show increased erythropoiesis, reduced erythroid cell apoptosis, and increased EPO mRNA and EPO levels.6,10,11 In vitro administration of iFGF23 to FGF23-knockout bone marrow–derived erythropoietic cells normalizes erythropoiesis, and administration of cFGF23 in chronic kidney disease (CKD) mice decreases erythroid cell apoptosis and increases serum EPO levels. Recombinant EPO stimulates a proportional increase in FGF23 mRNA transcription and iFGF23 cleavage, which translates into increased levels of circulating cFGF23 with only a modest increase in iFGF23 levels. Consequently, EPO alters the i:cFGF23 ratio in favor of cFGF23, overcoming the suppressive effects of iFGF23 on erythropoiesis.2-7 Thereby, iron deficiency and acute inflammation also decrease the i:cFGF23 ratio12-15 (Figure 1A; phase 2). On the other hand, an excess of circulating iFGF23 and a high i:cFGF23 ratio are observed in CKD as a consequence of impaired FGF23 cleavage in the presence of increased FGF23 transcription.16
In addition to FGF23, erythroferrone (ERFE) is a known hormone produced by erythroblasts in response to EPO, and it acts as a negative regulator of hepcidin during stress erythropoiesis.17 ERFE expression is expected to be increased in hereditary anemias with ineffective erythropoiesis (characterized by increased EPO levels), which will contribute to hepcidin suppression and subsequent iron loading. It is unknown whether ERFE interferes with or mediates EPO-FGF23 signaling.
As recently discussed,18 we hypothesize that EPO plays an important role in FGF23 signaling in hereditary hemolytic anemia. We explored this hypothesis in the current study, also taking into account ERFE, as a negative regulator of hepcidin.
Study design
Total FGF23, iFGF23, and EPO were measured in 90 patients and 10 healthy controls from the ZEbRA trial, a cross-sectional observational study on clinical sequelae and pathophysiology of rare congenital hemolytic diseases, which was conducted at the University Medical Center Utrecht (Netherlands Trial Register [NL5189]).
Laboratory parameters related to erythropoiesis, hemolysis, iron metabolism, kidney function, and calcium-phosphate metabolism were measured using routine laboratory procedures. An overview of relevant patient characteristics is provided in Table 1.
Two FGF23 enzyme-linked immunosorbent assays were used for FGF23 measurements in stored plasma samples: 1 that detects an epitope in the C-terminal part of the FGF23 molecule and, therefore, quantifies cFGF23 fragments and (full-length) iFGF23 (total FGF23; Immunotopics/Quidel, San Clemente, CA) and 1 that detects only iFGF23 (KAINOS Laboratories, Tokyo, Japan).
Plasma ERFE levels were quantified using an Intrinsic ERFE IE ELISA Kit (Intrinsic LifeSciences, San Diego, CA), according to the manufacturer’s instructions.
In addition, a broad range of chemokines and cytokines were measured (interleukin-1α [IL-1α], IL-1β, IL-2, IL-4, IL-5, IL-7, IL-8, IL-10, IL-12, IL-13, IL-15, IL-17, IL-21, IL-22, IL-23, IL-27, IL-31, tumor necrosis factor-α, tumor necrosis factor-β, interferon-γ, monocyte chemoattractant protein-1, macrophage inflammatory protein 1α [MIP-1α], MIP-1β, MIP-3α, eotaxin, monocyte chemoattractant protein-4, MDC, IP-10, PIGF, VEGF, soluble [s]CD163, HSP70, soluble vascular endothelial growth factor receptor 1, sICAM, and sVCAM) with a Luminex assay.
Correlations among total or iFGF23, ERFE, and EPO, as well as between iFGF23 and calcium, phosphate, 25-OH vitamin D, and CKD-EPI values (using the Chronic Kidney Disease Epidemiology Collaboration equation), were determined by linear regression analyses. In the case of parametric testing, bootstrapping was performed to confirm significance. Statistical significance for all tests was set at a 2-sided P < .05. Statistical analysis was performed using IBM SPSS software, version 25.0.0.2 (SPSS Inc., Chicago, IL).
The study was approved by the Medical Research Ethics Committee of the University Medical Center Utrecht in accordance with the Dutch Medical Research Involving Human Subjects Act and other applicable Dutch and European regulations.
Results and discussion
We observed a clear correlation between levels of total FGF23 and EPO in patients with various hereditary hemolytic anemias (r = +0.64, 95% confidence interval [CI], 0.09-0.89) (Figure 1B) that persisted after adjustment for iron load (transferrin saturation), inflammation (C-reactive protein), and kidney function (CKD-EPI). As expected, there was not a correlation between iFGF23 and EPO values or between iFGF23 and total FGF23. These data are consistent with a low i:cFGF23 ratio in response to EPO. To exclude a significant influence of calcium phosphate metabolism and kidney function on FGF23 regulation in our patients, we evaluated the relationship between biologically active FGF23 and serum calcium, phosphate, 25-OH vitamin D, and CKD-EPI measurements. iFGF23 did not correlate with serum calcium, phosphate, 25-OH vitamin D, or CKD-EPI (P > .05).
Although not an unexpected finding, we are the first to report a strong correlation between EPO and ERFE in a diverse set of patients with rare hereditary hemolytic anemias other than hemoglobinopathies,19-21 with and without transfusion dependency and with and without iron-reducing therapy (r = +0.47; 95% CI, 0.14-0.69). There was no correlation between ERFE and total FGF23 or iFGF23. ERFE is known to inhibit induction of hepcidin; hepcidin values were unavailable, which limited the ability to study the link between erythropoiesis and iron metabolism in our cohort. Hepcidin itself was previously shown to be capable of inducing FGF23 production and its cleavage, independently of inflammation.13 Our data underscore the importance of EPO-ERFE signaling in hemolytic anemia; however, ERFE is not simply an intermediary in EPO-FGF23 signaling. Our observation is in line with a previous study performed in mice.2 Thereby, we confirm the persistence of a relationship between EPO and FGF23 regulation in the absence of iron deficiency.
The influence of inflammation on the EPO-FGF23 axis22 was clearly illustrated by higher mean total FGF23 in patients with sickle cell disease (549 RU/L; standard deviation, 606 RU/L) compared with other patients (182 RU/L; standard deviation, 442 RU/L; mean difference, 367; 95% CI, 64-743), without a significant difference in EPO or iFGF23.
We identified a correlation between total FGF23 and MIP-1β ( = +0.49; 95% CI, 0.15-0.72) and between total FGF23 and sICAM (r = +0.54; 95% CI, 0.16-0.75), markers of endothelial dysfunction and immune players in pulmonary hypertension.23,24 Moreover, we speculate on the contribution of a decline in the i:cFGF23 ratio to vasculopathic complications. Our hypothesis is in line with previous observations that too little FGF signaling activates tumor growth factor-β signaling, leading to endothelial-to-mesenchymal transition and smooth muscle cell proliferation, which contribute to the development of pulmonary hypertension.25,26
In conclusion, we show, for the first time, a strong correlation between EPO and FGF23 signaling in a broad range of hereditary hemolytic anemias. Additionally, we show a clear, but not previously reported, correlation between EPO and ERFE in patients with a variety of hemolytic congenital anemias. These findings open up a new area for research and hold promise for potential future treatment in hereditary hemolytic anemia, because therapeutic targeting of the FGF23 pathway might provide an opportunity to ameliorate ineffective erythropoiesis and the development of complications.
Data sharing requests should be sent to Eduard J. van Beers (e.j.vanbeers-3@umcutrecht.nl).
Acknowledgment
The authors thank Wendy Dam for measuring iFGF23 and total FGF23 levels in plasma samples from the ZEbRA cohort.
Authorship
Contribution: S.v.S. assembled the cohort and prepared the samples for analysis; M.F.E., S.J.L.B., and M.H.d.B. facilitated FGF23 measurements; A.G. facilitated ERFE measurements; A.J.v.V. analyzed and interpreted the data and wrote the manuscript; R.v.W. and E.J.v.B. supervised the work; and all authors critically reviewed the manuscript and approved the final version for submission.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Eduard J. van Beers, Van Creveldkliniek UMC Utrecht, Huispost C01.428, Postbus 85500, 3508 GA Utrecht, The Netherlands; e-mail: e.j.vanbeers-3@umcutrecht.nl.