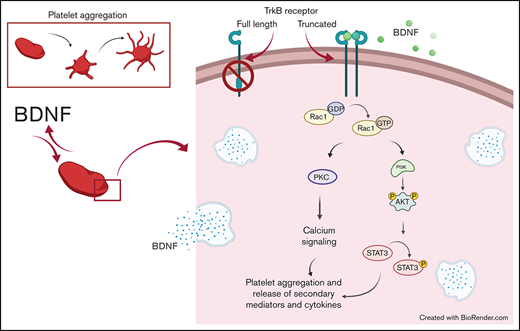
Key Points
Platelets release large amounts of BDNF, but its effect on platelet function is unknown.
We show BDNF to induce aggregation of washed human platelets through a truncated form of the TrkB receptor.

Abstract
Brain-derived neurotrophic factor (BDNF) has both autocrine and paracrine roles in neurons, and its release and signaling mechanisms have been extensively studied in the central nervous system. Large quantities of BDNF have been reported in circulation, essentially stored in platelets with concentrations reaching 100- to 1000-fold those of neurons. Despite this abundance, the function of BDNF in platelet biology has not been explored. At low concentrations, BDNF primed platelets, acting synergistically with classical agonists. At high concentrations, BDNF induced complete biphasic platelet aggregation that in part relied on amplification from secondary mediators. Neurotrophin-4, but not nerve growth factor, and an activating antibody against the canonical BDNF receptor tropomyosin-related kinase B (TrkB) induced similar platelet responses to BDNF, suggesting TrkB could be the mediator. Platelets expressed, both at their surface and in their intracellular compartment, a truncated form of TrkB lacking its tyrosine kinase domain. BDNF-induced platelet aggregation was prevented by inhibitors of Ras-related C3 botulinum toxin substrate 1 (Rac1), protein kinase C, and phosphoinositide 3-kinase. BDNF-stimulated platelets secreted a panel of angiogenic and inflammatory cytokines, which may play a role in maintaining vascular homeostasis. Two families with autism spectrum disorder were found to carry rare missense variants in the BDNF gene. Platelet studies revealed defects in platelet aggregation to low concentrations of collagen, as well as reduced adenosine triphosphate secretion in response to adenosine diphosphate. In summary, circulating BDNF levels appear to regulate platelet activation, aggregation, and secretion through activation of a truncated TrkB receptor and downstream kinase-dependent signaling.
Introduction
Initially discovered in the brain, brain-derived neurotrophic factor (BDNF) is a growth factor and a member of the neurotrophin family.1 BDNF has been extensively studied in the central nervous system and has a well-established role in synaptic plasticity and neuron development by promoting cell survival and neurite outgrowth.2,3 To exert its action, BDNF binds to the tropomyosin-related kinase B (TrkB) receptor, inducing receptor homodimerization and autophosphorylation within its endogenous kinase domain.4 Three TrkB isoforms have been reported, namely the full-length receptor and 2 truncated receptors (TrkB.T1 and TrkB.Shc in humans), that share the same extracellular domain but differ in their intracellular domains.5,6 The intracellular domains of TrkB.T1 and TrkB.Shc consist of a short cytoplasmic tail of 23 and 83 amino acids, respectively, and lack the intracellular tyrosine kinase domain of the full-length receptor.7 Notwithstanding, truncated TrkB receptors can signal through adaptor proteins or act as a dominant-negative receptor to inhibit BDNF signaling through the full-length TrkB receptor.8,9
TrkB receptors are found in many tissues outside of the central nervous system, including the lungs, heart, and vascular endothelium.10,11 Increasingly, BDNF is shown to play an important role in the cardiovascular system.11,12 In circulation, BDNF is stored primarily in platelets, where its concentrations can reach 100 to 1000 times those of the central nervous system.11,13 Interestingly, platelets have been shown to release BDNF upon activation, but the role of BDNF in platelets remains unknown.14 Since BDNF plays an autocrine-paracrine role in the brain, we hypothesized that BDNF would have a similar autocrine-paracrine role in platelets.15,16 We therefore sought to investigate the effect of BDNF on platelet function and intracellular signaling underlying platelet responses to BDNF. Here, we show that BDNF induces platelet aggregation by binding to a truncated TrkB receptor and activates a signaling pathway involving the Rho GTPase Ras-related C3 botulinum toxin substrate 1 (Rac1), protein kinase C (PKC), and phosphoinositide 3-kinase (PI3K). We also demonstrate that upon activation with BDNF, platelets release angiogenic and inflammatory cytokines, suggesting that BDNF may indeed play an autocrine-paracrine role in platelet function and vascular physiology.
Methods
A list of materials and more detailed methods can be found in supplemental Materials and methods.
Participant selection and blood collection
This study was approved by the Montreal Heart Institute Scientific and Research Ethics Committee (reference #2018-2368), and all participants gave written informed consent.
Blood was drawn by venipuncture into syringes containing acid citrate dextrose in a 1:5 volume ratio (acid citrate dextrose/blood) to prepare washed platelets.
Light transmission aggregometry
Platelet aggregation was measured using a ChronoLog aggregometer (Model 700 with AGGRO/LINK8 Software, Havertown, PA) at 37°C with continuous stirring at 1200 rpm. When specified, washed platelets were preincubated with inhibitors or vehicle for 15 minutes at room temperature.
Protein phosphorylation
To block the positive feedback from amplification pathways, washed platelets were preincubated for 15 minutes with eptifibatide (9 µM), aspirin (30 µM), and AR-C66096 (1 µM) prior to stimulation with agonists for 1 minute at 37°C under continuous stirring at 1200 rpm (ChronoLog model 700 aggregometer). Reactions were stopped with ice-cold RIPA buffer and proteins, resolved by SDS-PAGE on 8% acrylamide gels, and transferred onto polyvinylidene fluoride membranes for immunoblotting.
Flow cytometry and confocal microscopy
Cells and slides were prepared using standard protocols that are detailed in supplemental Materials and methods.
Platelet RNA sequencing
Platelet RNA sequencing from the publicly available thromboSeq RNA-sequencing pipeline was queried.17,18 Mapped read counts were selected for reads mapping to the TrkB gene (NTRK2; ENSG00000148053), BDNF (ENSG00000176697), and glycoprotein VI (GPVI; ENSG00000088053) and visualized in a dot plot. Counts per million were calculated using the R package edgeR employing the read counts summarized for all 57 736 annotated genes.
Analysis of the platelet secretome
Inflammatory and angiogenic profiles were assessed by multiplex bead kits (AimPlex Bioscience) and analyzed by flow cytometry (MACSQuant Analyzer 10, Miltenyi Biotec). Standard curves were generated and employed to convert the mean fluorescence intensity of each sample (run in duplicate) into concentration.
Identification of families with BDNF gene variants
Whole genomes from autism spectrum disorder (ASD) patients recruited by Leuven Autism Research19 were analyzed for rare variants in the BDNF gene as part of the NHIR BioResource rare disease study.20 Platelet aggregation and secretion assessments in these patients and their family members were carried out as previously reported.21 Plasma levels of BDNF were measured using an enzyme linked-immunosorbent assay (catalog no. BEK-2211, Biosensis). Presence of BDNF in platelet lysates was assessed by immunoblot analysis (BDNF antibody, catalog no. 500-P84, Peprotech; and rabbit polyclonal integrin β3 antibody, catalog no. sc-14009, Santa Cruz Biotechnology).
Statistical analyses
All data sets passed the Shapiro-Wilk test for normality. Repeated-measures analysis of variance (ANOVA) with Geisser-Greenhouse correction for sphericity and Dunnett’s correction for multiple comparisons was performed using GraphPad Prism Software version 8 for Windows (GraphPad Software, San Diego, CA). A multiplicity-adjusted P value < .05 was considered significant. Continuous variables are presented as median and interquartile range (IQR). N refers to the number of independent experiments with each experiment representing a different biological sample.
Results
BDNF induces platelet aggregation
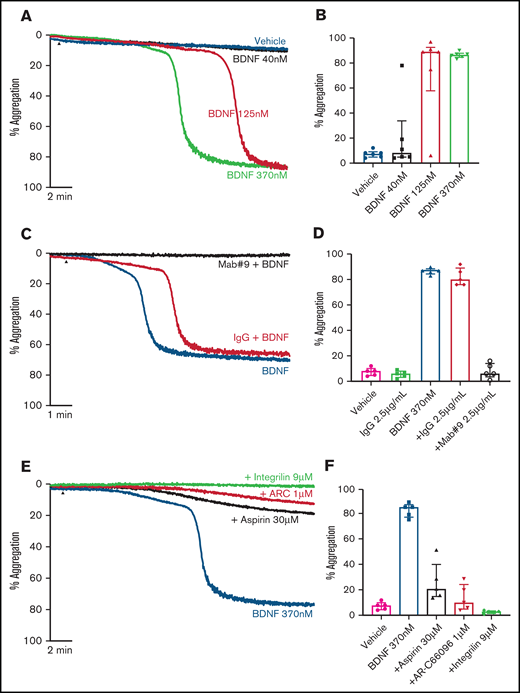
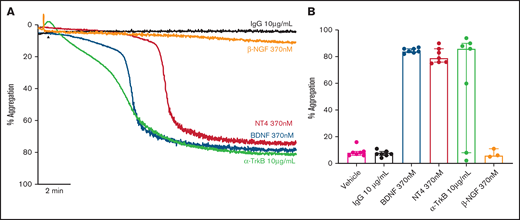
Addition of BDNF (1, 3, and 10 µg/mL equivalent to 40 nM, 125 nM, and 370 nM) induced platelet aggregation in a concentration-dependent manner (Figure 1A-B). This aggregation was confirmed with BDNF sourced from different vendors, as shown in supplemental Figure 1. Although BDNF induced complete and irreversible aggregation, the lag time between agonist addition and initiation of aggregation was variable between individuals (supplemental Figure 2). In the presence of a neutralizing antibody against BDNF (mab #9), but not an isotype immunoglobulin G (IgG) control, platelet aggregation in response to BDNF was prevented (Figure 1C-D). To further confirm that BDNF induced platelet aggregation and not agglutination, we investigated the contribution of amplification pathways to BDNF-induced platelet aggregation. Aspirin (COX-1 inhibition), AR-C66096 (ADP-P2Y12 receptor inhibition), and eptifibatide (integrin α2bβ3 inhibition) significantly impaired the ability of BDNF to induce platelet aggregation (Figure 1E-F).
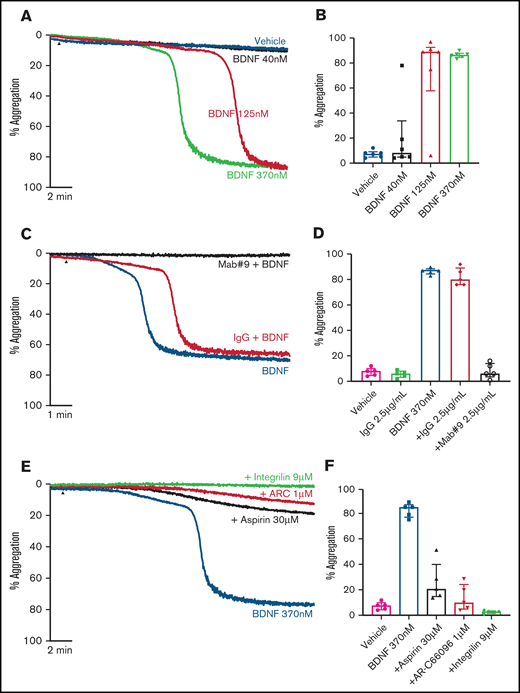
BDNF induces complete, biphasic platelet aggregation in washed platelets. Representative trace (A) and quantification (B) of platelet aggregation in response to 40, 125, and 370 nM BDNF. Repeated-measures ANOVA, P = .0004; vehicle vs 125 nM BDNF, P = .01; vehicle vs BDNF 370 nM, P < .0001. Example trace (C) and quantification (D) of neutralization of BDNF-induced aggregation by the mab#9 antibody (2.5 µg/mL). IgG2B was used as an isotype control (2.5 µg/mL). Results are representative of ≥5 independent experiments. Repeated-measures ANOVA, P < .0001; BDNF 370 nM vs BDNF 370 nM in the presence of mab#9, P < .0001. Example trace (E) and quantification (F) of BDNF-induced platelet aggregation in the presence of inhibitors of secondary mediators (30 µM aspirin, 1 µM AR-C66096 (ARC), and 9 µM eptifibatide (Integrilin)). Results are representative of 5 independent experiments. Repeated-measures ANOVA, P < .0001; compared with BDNF 370 nM: inhibition with aspirin, P = .001; inhibition with AR-C66096, P = .0002; and inhibition with eptifibatide, P < .0001. Arrowheads indicate the time point at which the agonist was added. Data are presented as median and IQR.
BDNF induces complete, biphasic platelet aggregation in washed platelets. Representative trace (A) and quantification (B) of platelet aggregation in response to 40, 125, and 370 nM BDNF. Repeated-measures ANOVA, P = .0004; vehicle vs 125 nM BDNF, P = .01; vehicle vs BDNF 370 nM, P < .0001. Example trace (C) and quantification (D) of neutralization of BDNF-induced aggregation by the mab#9 antibody (2.5 µg/mL). IgG2B was used as an isotype control (2.5 µg/mL). Results are representative of ≥5 independent experiments. Repeated-measures ANOVA, P < .0001; BDNF 370 nM vs BDNF 370 nM in the presence of mab#9, P < .0001. Example trace (E) and quantification (F) of BDNF-induced platelet aggregation in the presence of inhibitors of secondary mediators (30 µM aspirin, 1 µM AR-C66096 (ARC), and 9 µM eptifibatide (Integrilin)). Results are representative of 5 independent experiments. Repeated-measures ANOVA, P < .0001; compared with BDNF 370 nM: inhibition with aspirin, P = .001; inhibition with AR-C66096, P = .0002; and inhibition with eptifibatide, P < .0001. Arrowheads indicate the time point at which the agonist was added. Data are presented as median and IQR.
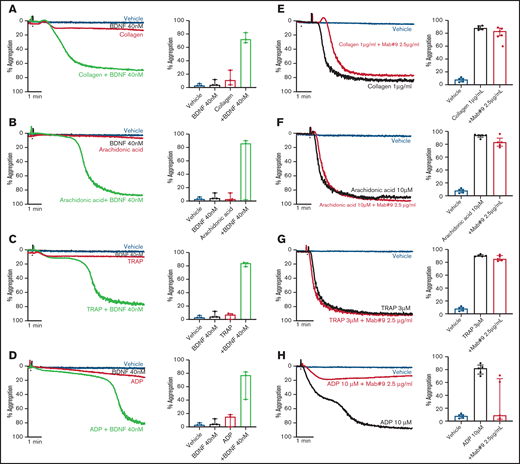
To investigate possible priming/synergistic effects of BDNF on platelet aggregation, we preincubated platelets with 40 nM BDNF for 15 minutes prior to addition of subthreshold concentrations of classical platelet agonists. Preincubation with BDNF led to complete and irreversible aggregation with subthreshold concentrations of collagen, arachidonic acid, thrombin receptor activating peptide (TRAP), and adenosine diphosphate (ADP) (Figure 2A-D).
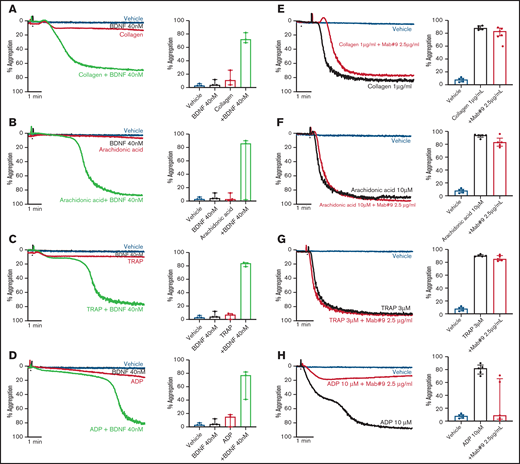
BDNF acts as a platelet primer to other classical agonists. Example trace and quantification of platelet aggregation of platelets pre-incubated with BDNF (40 nM) in response to subthreshold concentrations of collagen (A) (0.125-0.5 µg/mL; repeated-measures ANOVA test, P = .01; compared with vehicle, both BDNF and collagen, P > .05; compared with collagen alone, collagen + BDNF, P = .005), arachidonic acid (B) (0.5-2.5 µM; repeated-measures ANOVA test, P = .208), TRAP (C) (0.5 µM; repeated-measures ANOVA test, P = .0002; compared with vehicle, both BDNF and TRAP, P > .05; compared with TRAP alone, TRAP + BDNF, P = .008), and ADP (D) (0.5 µM; repeated-measures ANOVA test, P = .03; compared with vehicle, both BDNF and ADP, P > .05; compared with ADP alone, ADP + BDNF, P = .07). Example trace and quantification of platelet aggregation in the presence of the BDNF-neutralizing antibody (mab#9) in response to 1 µg/mL collagen (E) (repeated-measures ANOVA, P < .0001; compared with vehicle, collagen alone and/or in presence of mab#9, P < .0001; compared with collagen alone, collagen in the presence of mab#9, P > .05), 10 µM arachidonic acid (F) (repeated-measures ANOVA, P < .0001; compared with vehicle, arachidonic acid alone and/or in the presence of mab#9, P < .0001; compared with arachidonic acid alone, arachidonic acid in the presence of mab#9, P > .05); 3 µM TRAP (G) (repeated-measures ANOVA, P < .0001; compared with vehicle, TRAP alone and/or in the presence of mab#9, P < .0001; compared with TRAP alone, TRAP in the presence of mab#9, P > .05); and 10 µM ADP (H) (repeated-measures ANOVA, P = .01; compared with vehicle, ADP alone, P < .0001; compared with ADP in the presence of mab#9, both vehicle and ADP alone, P > .05). Arrowheads indicate the time point at which the agonist was added. Data are presented as median and IQR.
BDNF acts as a platelet primer to other classical agonists. Example trace and quantification of platelet aggregation of platelets pre-incubated with BDNF (40 nM) in response to subthreshold concentrations of collagen (A) (0.125-0.5 µg/mL; repeated-measures ANOVA test, P = .01; compared with vehicle, both BDNF and collagen, P > .05; compared with collagen alone, collagen + BDNF, P = .005), arachidonic acid (B) (0.5-2.5 µM; repeated-measures ANOVA test, P = .208), TRAP (C) (0.5 µM; repeated-measures ANOVA test, P = .0002; compared with vehicle, both BDNF and TRAP, P > .05; compared with TRAP alone, TRAP + BDNF, P = .008), and ADP (D) (0.5 µM; repeated-measures ANOVA test, P = .03; compared with vehicle, both BDNF and ADP, P > .05; compared with ADP alone, ADP + BDNF, P = .07). Example trace and quantification of platelet aggregation in the presence of the BDNF-neutralizing antibody (mab#9) in response to 1 µg/mL collagen (E) (repeated-measures ANOVA, P < .0001; compared with vehicle, collagen alone and/or in presence of mab#9, P < .0001; compared with collagen alone, collagen in the presence of mab#9, P > .05), 10 µM arachidonic acid (F) (repeated-measures ANOVA, P < .0001; compared with vehicle, arachidonic acid alone and/or in the presence of mab#9, P < .0001; compared with arachidonic acid alone, arachidonic acid in the presence of mab#9, P > .05); 3 µM TRAP (G) (repeated-measures ANOVA, P < .0001; compared with vehicle, TRAP alone and/or in the presence of mab#9, P < .0001; compared with TRAP alone, TRAP in the presence of mab#9, P > .05); and 10 µM ADP (H) (repeated-measures ANOVA, P = .01; compared with vehicle, ADP alone, P < .0001; compared with ADP in the presence of mab#9, both vehicle and ADP alone, P > .05). Arrowheads indicate the time point at which the agonist was added. Data are presented as median and IQR.
Next, we assessed whether the BDNF-neutralizing antibody inhibited platelet aggregation responses to higher agonist concentrations. Whereas the BDNF-neutralizing antibody did not inhibit final aggregation to collagen (Figure 2E), the lag time to induce aggregation in response to collagen was prolonged (43 ± 11 seconds vs 57 ± 11 seconds in the presence of mab#9, P < .05). Aggregation responses to arachidonic acid and TRAP were maintained in the presence of the BDNF-neutralizing antibody (Figure 2F-G). Conversely, ADP-induced platelet aggregation was inhibited by preincubation with the BDNF-neutralizing antibody, although the extent of inhibition was highly variable (Figure 2H).
Taken together, these findings show that BDNF induces platelet aggregation through an active mechanism that may prime platelets to other agonists and, in part, relies on amplification from secondary mediators.
Platelets express a truncated form of the TrkB receptor
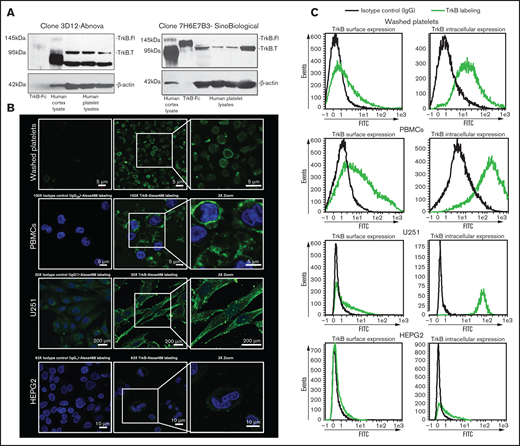
We investigated the presence of the canonical BDNF receptor, TrkB, on washed platelets with 2 independent antibodies. The specificity of the antibodies was verified by competition assays with immunizing peptides (supplemental Figure 3). Whereas both the full-length receptor (145 kDa) and the truncated receptor (95 kDa) were present in the adult human brain cerebral cortex whole-tissue lysate (5 µg loaded, used as a positive control), only the truncated receptor was detected in platelet lysates (100 µg loaded; Figure 3A). We then examined if the NTRK2 gene, coding for TrkB, was translated in human platelets by RNA sequencing. NTRK2 mRNA was detectable only in a small subset of platelets of healthy individuals and at very low counts; in contrast, BDNF was detected in a significantly larger subset of samples, and GPVI was detected in nearly all samples (supplemental Figure 4). These data suggest that although platelets may express TrkB, the expression level is likely to be very low, which is consistent with the results from immunoblotting assays.
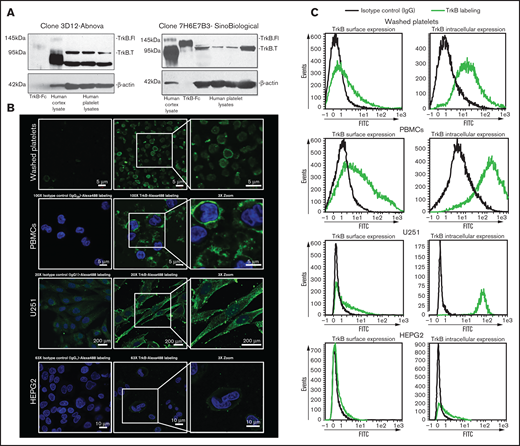
Platelets express truncated TrkB receptors. (A) Immunoblotting of TrkB from human platelet lysates (100 µg for Sino Biological clone 7H6E7B3 and 25 µg for Abnova clone 3D12) obtained from 4 different healthy volunteers. TrkB-Fc fusion protein (5 ng; expected molecular weight, 120 kDa) and human cortex whole cell lysate (5 µg; expected molecular weight of truncated TrkB and the full-length TrkB receptor, 95- and 140-kDa, respectively) were used as positive controls. β-Actin was used a loading control. Two different antibodies (clone 7H6E7B3 and clone 3D12) targeting the extracellular domain of TrkB were used. Blots are representative of ≥3 independent experiments. (B) Confocal fluorescence microscopy of platelets expressing TrkB. Washed platelets, PBMCs, U251-MG cells, and HEPG2 cells were labeled with anti-TrkB primary antibodies and Alexa Fluor 488–conjugated secondary antibodies. Nuclei of PBMCs, U251-MG cells, and HEPG2 cells were stained with 4′,6-diamidino-2-phenylindole, and IgG2B/IgG1 was used as an isotype control. Images were visualized at room temperature with Zeiss LSM510 using a 100× objective lens for platelets and PBMCs, a 63× objective lens for HEPG2 cells, and a 20× objective lens for U251-MG cells as well as 3× magnification. Scale bar represents 5 µm for PBMCs and platelets, 10 µm for HEPG2 cells, and 200 µm for U251-MG cells. Images are representative of 3 independent experiments. (C) Flow cytometry of surface and intracellular TrkB on washed human platelets; platelets expressed TrkB on both their surface (TrkB+: 27% ± 11%, n = 8) and their intracellular compartment (TrkB+: 82% ± 9%, n = 6). PBMCs were used as positive controls for TrkB labeling on both the surface (TrkB+: 45% ± 15%, n = 4) and the intracellular compartment (TrkB+: 88% ± 6%, n = 4). U251-MG cells were used as positive controls for TrkB labeling on both the surface (TrkB+: 30% ± 2%, n = 3) and the intracellular compartment (TrkB+: 94% ± 2%, n = 3). IgG1/2B was used as isotype control. HEPG2 cells were used as TrkB-low controls for TrkB labeling on both the surface (TrkB+: 6% ± 0.12%, n = 2) and the intracellular compartment (TrkB+: 17% ± 1%, n = 2). FITC, fluorescein isothiocyanate.
Platelets express truncated TrkB receptors. (A) Immunoblotting of TrkB from human platelet lysates (100 µg for Sino Biological clone 7H6E7B3 and 25 µg for Abnova clone 3D12) obtained from 4 different healthy volunteers. TrkB-Fc fusion protein (5 ng; expected molecular weight, 120 kDa) and human cortex whole cell lysate (5 µg; expected molecular weight of truncated TrkB and the full-length TrkB receptor, 95- and 140-kDa, respectively) were used as positive controls. β-Actin was used a loading control. Two different antibodies (clone 7H6E7B3 and clone 3D12) targeting the extracellular domain of TrkB were used. Blots are representative of ≥3 independent experiments. (B) Confocal fluorescence microscopy of platelets expressing TrkB. Washed platelets, PBMCs, U251-MG cells, and HEPG2 cells were labeled with anti-TrkB primary antibodies and Alexa Fluor 488–conjugated secondary antibodies. Nuclei of PBMCs, U251-MG cells, and HEPG2 cells were stained with 4′,6-diamidino-2-phenylindole, and IgG2B/IgG1 was used as an isotype control. Images were visualized at room temperature with Zeiss LSM510 using a 100× objective lens for platelets and PBMCs, a 63× objective lens for HEPG2 cells, and a 20× objective lens for U251-MG cells as well as 3× magnification. Scale bar represents 5 µm for PBMCs and platelets, 10 µm for HEPG2 cells, and 200 µm for U251-MG cells. Images are representative of 3 independent experiments. (C) Flow cytometry of surface and intracellular TrkB on washed human platelets; platelets expressed TrkB on both their surface (TrkB+: 27% ± 11%, n = 8) and their intracellular compartment (TrkB+: 82% ± 9%, n = 6). PBMCs were used as positive controls for TrkB labeling on both the surface (TrkB+: 45% ± 15%, n = 4) and the intracellular compartment (TrkB+: 88% ± 6%, n = 4). U251-MG cells were used as positive controls for TrkB labeling on both the surface (TrkB+: 30% ± 2%, n = 3) and the intracellular compartment (TrkB+: 94% ± 2%, n = 3). IgG1/2B was used as isotype control. HEPG2 cells were used as TrkB-low controls for TrkB labeling on both the surface (TrkB+: 6% ± 0.12%, n = 2) and the intracellular compartment (TrkB+: 17% ± 1%, n = 2). FITC, fluorescein isothiocyanate.
To determine the subcellular distribution of the receptor, surface staining and intracellular staining for TrkB was assessed by confocal microscopy (Figure 3B) and flow cytometry (Figure 3C) in fixed and permeabilized platelets. Up to 77% of platelets were TrkB+ by flow cytometry, and TrkB immunoreactivity was observed at the plasma membrane and distributed in a punctuate manner in the cytoplasm of washed platelets, suggesting a granular compartmentalization. Similar TrkB immunoreactivity was observed with peripheral blood mononuclear cells (PBMCs) and the U-251 MG glioblastoma cells used as TrkB-high controls in comparison with HEPG2 hepatoma cells used as TrkB-low controls.
Other ligands of TrkB also induce platelet aggregation
We assessed whether other known ligands of the TrkB receptor induced platelet aggregation in a manner similar to BDNF. Both neurotrophin-4 (NT-4; 10 µg/mL equivalent to 370 nM) and a TrkB-activating antibody (clone 7H6E7B3, 10 µg/mL) induced complete, biphasic platelet aggregation (Figure 4A-B), further suggesting TrkB could be the target receptor in platelets. Nerve growth factor (NGF), a protein with similar physicochemical properties to BDNF and NT-4 but acting through the TrkA receptor, failed to elicit platelet aggregation (Figure 4A-B).
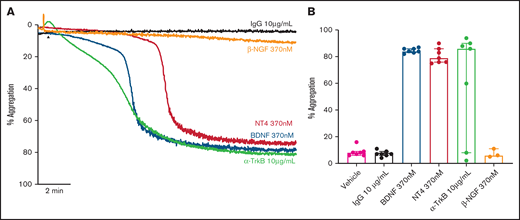
Known ligands of TrkB induce platelet aggregation. Example trace (A) and quantification (B) of platelet aggregation in response to 370 nM BDNF, 370 nM NT4, 10 µg/ml TrkB-activating antibody (n = 7), and 370 nM β-NGF (n = 3). IgG1 was used as an isotype control (10 µg/mL). Repeated-measures ANOVA, P = .005; compared with vehicle, BDNF 370 nM P < .0001, NT4 370 nM P < .0001 and TrkB-activating antibody, P = .037. Arrowheads indicate the time point at which the agonist was added. Data are presented as median and IQR.
Known ligands of TrkB induce platelet aggregation. Example trace (A) and quantification (B) of platelet aggregation in response to 370 nM BDNF, 370 nM NT4, 10 µg/ml TrkB-activating antibody (n = 7), and 370 nM β-NGF (n = 3). IgG1 was used as an isotype control (10 µg/mL). Repeated-measures ANOVA, P = .005; compared with vehicle, BDNF 370 nM P < .0001, NT4 370 nM P < .0001 and TrkB-activating antibody, P = .037. Arrowheads indicate the time point at which the agonist was added. Data are presented as median and IQR.
BDNF induces platelet aggregation through a kinase-dependent mechanism
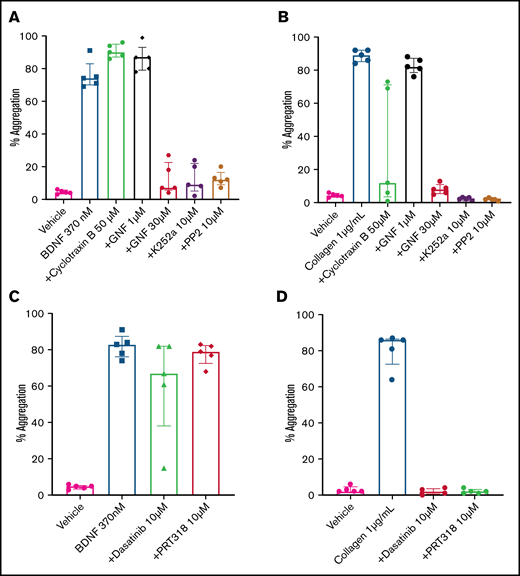
The immunoblotting results suggested that platelets express a 95-kDa isoform of the TrkB receptor, which corresponds to the expected mass for a truncated TrkB receptor lacking its kinase domain.7 In order to confirm that BDNF-induced platelet aggregation was not dependent on an intrinsic TrkB tyrosine kinase signaling domain, we used cyclotraxin B, an allosteric modulator of the TrkB receptor that induces a conformational change preventing autophosphorylation of the TrkB kinase domain,22 and GNF5837, an inhibitor of TrkB kinase activity that is specific to TrkB in the nanomolar range but loses specificity above 1 µM.23 Concentrations of cyclotraxin B up to 50 µM failed to inhibit BDNF-induced platelet aggregation, although it did inhibit collagen-induced aggregation in a subset of participants (Figure 5A). Incubation of platelets with a low concentration of GNF5837 (1 µM) did not impair platelet aggregation in response to BDNF (Figure 5A). At 30 µM, GNF5837 significantly decreased BDNF-induced platelet aggregation, as well as that of the collagen control (Figure 5A-B).
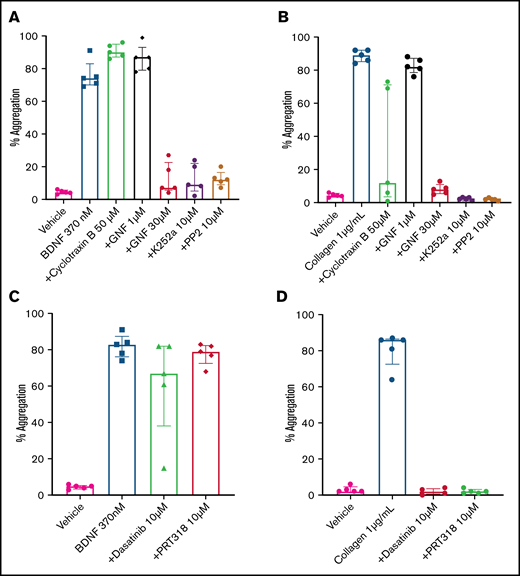
BDNF-induced aggregation activates a kinase-dependent pathway. (A) BDNF-induced platelet aggregation in the presence of TrkB kinase domain inhibitors (cyclotraxin B 50 µM and GNF5837 1 µM and 30 µM) as well as broad-spectrum kinase inhibitors (K252a 10 µM and PP2 10 µM). (B) Collagen at 1 µg/mL was used as control (n = 5). Repeated-measures ANOVA, P < .0001; compared with BDNF: no inhibition observed with cyclotraxin B 50 µM and GN5837 1 µM, P > .05; inhibition of BDNF with GNF5837 30 µM, P = .0025; inhibition with K252a 10 µM, P = .004; and inhibition with PP2 10 µM, P = .001. (C) BDNF-induced aggregation in the presence of SFK inhibitors (PRT318 10 µM and dasatinib 10 µM; n = 5). (D) Collagen at 1 µg/mL was used as control (n = 5). Repeated-measures ANOVA, P = .23; inhibition of BDNF with dasatinib 10 µM, P = .34; inhibition with PRT318 10 µM, P = .20. Data are presented as median and IQR.
BDNF-induced aggregation activates a kinase-dependent pathway. (A) BDNF-induced platelet aggregation in the presence of TrkB kinase domain inhibitors (cyclotraxin B 50 µM and GNF5837 1 µM and 30 µM) as well as broad-spectrum kinase inhibitors (K252a 10 µM and PP2 10 µM). (B) Collagen at 1 µg/mL was used as control (n = 5). Repeated-measures ANOVA, P < .0001; compared with BDNF: no inhibition observed with cyclotraxin B 50 µM and GN5837 1 µM, P > .05; inhibition of BDNF with GNF5837 30 µM, P = .0025; inhibition with K252a 10 µM, P = .004; and inhibition with PP2 10 µM, P = .001. (C) BDNF-induced aggregation in the presence of SFK inhibitors (PRT318 10 µM and dasatinib 10 µM; n = 5). (D) Collagen at 1 µg/mL was used as control (n = 5). Repeated-measures ANOVA, P = .23; inhibition of BDNF with dasatinib 10 µM, P = .34; inhibition with PRT318 10 µM, P = .20. Data are presented as median and IQR.
K252a (10 µM), an inhibitor of protein serine/threonine and protein tyrosine kinases, blocked BDNF- and collagen-induced platelet aggregation (Figure 5A-B), as did PP2 (10 µM), a nonselective inhibitor of the Src family of tyrosine kinases (SFKs) (Figure 5A-B). Taken together, these findings suggest that BDNF signals through a truncated form of the TrkB receptor that is devoid of its tyrosine kinase domain but may engage adaptor proteins to signal through one or more non-receptor protein kinase–dependent pathways.
BDNF-induced platelet responses do not require activation of non-receptor protein tyrosine kinases Src and Syk
Given the parallels with collagen-induced platelet aggregation, we next investigated whether protein tyrosine kinases Src and Syk were involved in BDNF-induced platelet aggregation similar to the collagen-induced activation of GPVI.24,25 In contrast to collagen, the Src-specific inhibitor dasatinib (10 µM) and the Syk-specific inhibitor PRT318 (10 µM) failed to inhibit BDNF-induced platelet aggregation (Figure 5C-D), suggesting that different protein tyrosine kinases are involved in platelet responses to BDNF.
Rho GTPase Rac-1 is involved in BDNF-induced platelet aggregation
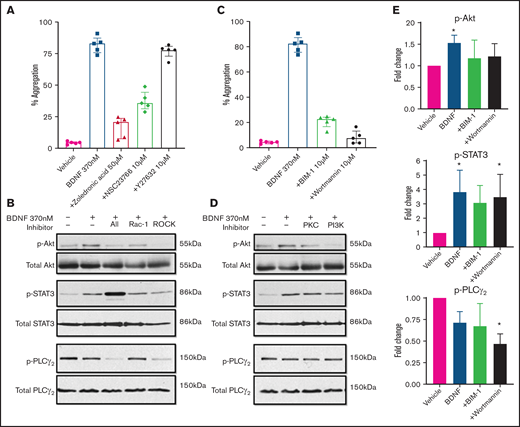
As ras homologous (Rho) GTPases have been shown to mediate truncated TrkB signaling in astrocytes, glioma, and glial cells,26,27 we investigated their possible role in BDNF-induced platelet aggregation. Zoledronic acid (50 µM), a nonspecific Rho inhibitor, significantly inhibited BDNF-induced platelet aggregation (Figure 6A). We next examined the effects of targeted inhibition of Rho family members Rac-1 and ras homologous-associated protein kinase (ROCK), a major downstream effector of rhoA. The Rac-1 inhibitor NSC23766 (10 µM) decreased platelet aggregation in response to BDNF, whereas the ROCK inhibitor Y27632 (10 µM) had no effect (Figure 6A).28,29 Collagen-induced aggregation was variably affected by inhibition of Rho GTPases (supplemental Figure 5A), as previously described.30
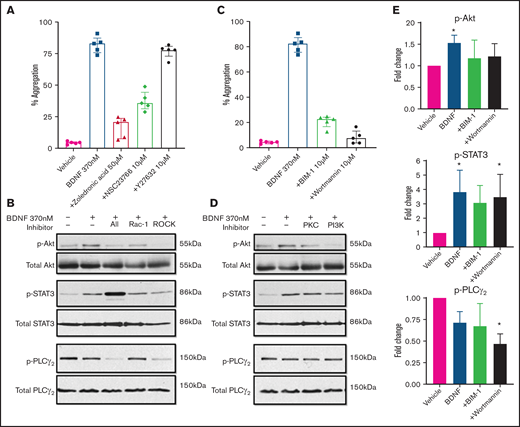
BDNF-induced aggregation recruits Rho GTPase Rac1 and activates PKC and PI3K/Akt pathway. (A) BDNF-induced aggregation in the presence of Rho GTPase inhibitors (zoledronic acid 50 µM, NSC23766 10 µM, and Y27632 10 µM; n = 5). Repeated-measures ANOVA, P < .0001; inhibition of BDNF with zoledronic acid, P = .0002; inhibition with NSC23766, P = .005; and inhibition with Y27632, P = .53. (B) BDNF-induced phosphorylation of Akt, STAT3, and PLC-γ2 in the presence of Rho GTPase inhibitors (zoledronic acid 50 µM, NSC23766 10 µM, and Y27632 10 µM). (C) BDNF-induced aggregation in the presence of PI3K and PKC inhibitors (wortmannin 100 nM and BIM-1 10 µM, n = 5). Repeated-measures ANOVA, P < .0001; inhibition of BDNF with BIM-1, P < .0001, inhibition with wortmannin, P < .0001. (D) BDNF-induced phosphorylation of Akt, STAT3, and PLC-γ2 in the presence of PI3K and PKC inhibitors (wortmannin 100 nM and BIM-1 10 µM). (E) Quantification of BDNF-induced phosphorylation of Akt, STAT3, and PLC-γ2 in the presence of PI3K and PKC inhibitors (wortmannin 100 nM and BIM-1 10 µM). One-way ANOVA; compared with vehicle: phosphorylation of Akt by BDNF is increased (P < .05), and there is no significant difference in the presence of BIM-1 and wortmannin. Phosphorylation of STAT3 by BDNF is increased (P < .05) with BDNF in the presence of wortmannin (P < .05), and there is no significant difference with BIM-1. Phosphorylation of PLC-γ2 by BDNF in the presence of wortmannin, P < .05; no significant difference with BDNF alone or in the presence of BIM-1. Density was measured with ImageJ and is expressed as a ratio of density of phosphorylated protein/density of total protein and standardized to the vehicle control. Functional data and phosphoblots are representative of ≥3 independent experiments.
BDNF-induced aggregation recruits Rho GTPase Rac1 and activates PKC and PI3K/Akt pathway. (A) BDNF-induced aggregation in the presence of Rho GTPase inhibitors (zoledronic acid 50 µM, NSC23766 10 µM, and Y27632 10 µM; n = 5). Repeated-measures ANOVA, P < .0001; inhibition of BDNF with zoledronic acid, P = .0002; inhibition with NSC23766, P = .005; and inhibition with Y27632, P = .53. (B) BDNF-induced phosphorylation of Akt, STAT3, and PLC-γ2 in the presence of Rho GTPase inhibitors (zoledronic acid 50 µM, NSC23766 10 µM, and Y27632 10 µM). (C) BDNF-induced aggregation in the presence of PI3K and PKC inhibitors (wortmannin 100 nM and BIM-1 10 µM, n = 5). Repeated-measures ANOVA, P < .0001; inhibition of BDNF with BIM-1, P < .0001, inhibition with wortmannin, P < .0001. (D) BDNF-induced phosphorylation of Akt, STAT3, and PLC-γ2 in the presence of PI3K and PKC inhibitors (wortmannin 100 nM and BIM-1 10 µM). (E) Quantification of BDNF-induced phosphorylation of Akt, STAT3, and PLC-γ2 in the presence of PI3K and PKC inhibitors (wortmannin 100 nM and BIM-1 10 µM). One-way ANOVA; compared with vehicle: phosphorylation of Akt by BDNF is increased (P < .05), and there is no significant difference in the presence of BIM-1 and wortmannin. Phosphorylation of STAT3 by BDNF is increased (P < .05) with BDNF in the presence of wortmannin (P < .05), and there is no significant difference with BIM-1. Phosphorylation of PLC-γ2 by BDNF in the presence of wortmannin, P < .05; no significant difference with BDNF alone or in the presence of BIM-1. Density was measured with ImageJ and is expressed as a ratio of density of phosphorylated protein/density of total protein and standardized to the vehicle control. Functional data and phosphoblots are representative of ≥3 independent experiments.
We then investigated the involvement of PKC and the PI3K/protein kinase B (Akt) pathway, which are major signaling pathways in platelets and neurons and have been reported to be activated downstream of TrkB in the central nervous system. Inhibition of either PKC (BIM-1, 10 µM) or PI3K (wortmannin, 100 nM) significantly attenuated both BDNF-induced (Figure 6C) and collagen-induced (supplemental Figure 5C) platelet aggregation. It should be noted that at the concentration used in the present study, BIM-1 would also inhibit GSK3 activity.
We further confirmed the aggregation results by immunoblotting for the phosphorylated form of relevant proteins. As shown in Figure 6B, Rho GTPase inhibition reduced BDNF-induced Akt phosphorylation and the signal transducer and activator of transcription 3 protein (STAT3) phosphorylation, while PLCγ2 phosphorylation was not affected. PI3K and PKC inhibitors (wortmannin and BIM-1) both decreased BDNF-induced phosphorylation of Akt and STAT3, but not PLCγ2, as shown in Figure 6D-E. Collagen-induced phosphorylation, used as control, in the presence of PI3K/PKC inhibitors is shown in supplemental Figure 5B,D-E. Taken together, these results suggest BDNF induces aggregation through recruitment of Rac1 as an adaptor protein that induces activation of PKC and the PI3K pathway and phosphorylation of STAT3. While PLCγ2 is not involved in BDNF signaling, PKC and PI3K appear to act through pathways independent of STAT3.
BDNF induces platelet secretion
We next investigated the possibility that BDNF plays an autocrine/paracrine role in a growing thrombus by inducing platelet secretion. BDNF (370 nM) induced the release of several inflammatory and angiogenic factors, such as interleukin-8, ENA-78, TARC, and VEGF-A (supplemental Figure 6). The release reaction was similar to that induced by collagen-related peptide, TRAP, and PAR-4 amide.
Rare genetic BDNF missense variants are associated with autism and platelet function defects
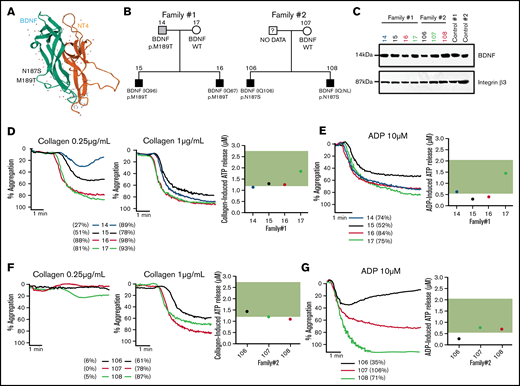
As reduced BDNF blood levels and BDNF haploinsufficiency have previously been associated with autism, we have analyzed a unique cohort of ASD patients from whom platelet function data and whole genomes were already available for rare genetic variants in BDNF.20,21,31,32 Two families from these 57 ASD families were found to carry rare heterozygous BDNF missense variants (N187S and M189T), both predicted to be pathogenic, with combined annotation-dependent depletion scores of 23 and 16 and a minor allele frequency in the gnomAD population database of 0.0000144 and 0.0000039, respectively. These variants are located in loop III of the mature BDNF protein (Figure 7A), which is important in mediating BDNF-TrkB interactions.33,34 Index patients, their siblings, and parents (Figure 7B) were assessed.
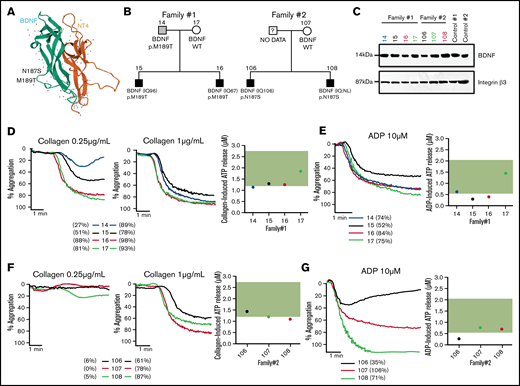
BDNF N187S and M189T variants influence platelet function. (A) Modeled structure of BDNF and NT4. BDNF is represented in green and NT4 in orange. The arrows represent the B-strands, and the coiled regions are represented by the thin lines. N187S and M189T are indicated on the 3D structure of BDNF and are located on loop III. (B) Family trees of the 2 families carrying the rare BDNF variants. (C) Immunoblotting of BDNF in platelet lysates of the 2 ASD families; 50 µg of protein was loaded in each well, and 2 healthy volunteers were used as controls. Integrin β3 was used as a loading control. For carriers of BDNF N187S variant, traces of platelet aggregation in response to 0.25 µg/mL and 1 µg/mL collagen are shown, as well as ATP release in response to 2 µg/mL collagen (D). (E) Traces of platelet aggregation as well as ATP release in response to 10 µM of ADP. For carriers of BDNF M189T variant, traces of platelet aggregation in response to 0.25 µg/mL and 1 µg/mL collagen are shown, as well as ATP release in response to 2 µg/mL collagen (F). (G) Traces of platelet aggregation as well as ATP release in response to 10 µM of ADP. The green band corresponds to the local normal reference range for ATP secretion (n = 40). Participants are color-coded; their maximal platelet aggregation (%) in platelet-rich plasma is indicated in the legend.
BDNF N187S and M189T variants influence platelet function. (A) Modeled structure of BDNF and NT4. BDNF is represented in green and NT4 in orange. The arrows represent the B-strands, and the coiled regions are represented by the thin lines. N187S and M189T are indicated on the 3D structure of BDNF and are located on loop III. (B) Family trees of the 2 families carrying the rare BDNF variants. (C) Immunoblotting of BDNF in platelet lysates of the 2 ASD families; 50 µg of protein was loaded in each well, and 2 healthy volunteers were used as controls. Integrin β3 was used as a loading control. For carriers of BDNF N187S variant, traces of platelet aggregation in response to 0.25 µg/mL and 1 µg/mL collagen are shown, as well as ATP release in response to 2 µg/mL collagen (D). (E) Traces of platelet aggregation as well as ATP release in response to 10 µM of ADP. For carriers of BDNF M189T variant, traces of platelet aggregation in response to 0.25 µg/mL and 1 µg/mL collagen are shown, as well as ATP release in response to 2 µg/mL collagen (F). (G) Traces of platelet aggregation as well as ATP release in response to 10 µM of ADP. The green band corresponds to the local normal reference range for ATP secretion (n = 40). Participants are color-coded; their maximal platelet aggregation (%) in platelet-rich plasma is indicated in the legend.
Plasma BDNF levels were within the normal range (1003-2716 pg/mL) for carriers of the M189T variant (1297 ± 261 pg/mL) but lower than in controls in carriers of the N187S variant (869 ± 98 pg/mL) (Table 1). It should be noted, however, that BDNF levels were low in the mother (noncarrier) of the patients with the N187S variant (669 pg/mL); thus, the variant may not be the only determinant of BDNF levels in this family. BDNF was detectable in platelet lysates of all tested participants, regardless of genotype (Figure 7C).
Platelet studies revealed defects in platelet aggregation at low collagen concentrations, while at high collagen concentrations, platelet aggregation was similar between carriers and noncarriers for the M189T variant; the extent of impairment varied between individuals (Figure 7D). All participants of the family carrying the N187S mutation had low circulating BDNF levels (Table 1) and impaired platelet aggregation responses to low-concentration collagen (Figure 7F), regardless of genotype. Adenosine triphosphate (ATP) release after platelet activation with a high collagen concentration (2 µg/mL) was at the low end or slightly below the reference range from healthy volunteers (Figure 7D,F), regardless of BDNF variant.
In addition to impairments in response to collagen, platelet aggregation responses to ADP, even at a high concentration, were variable among affected individuals. Moreover, ATP secretion in response to ADP was reduced in carriers of BDNF variants, even when aggregation to ADP was preserved (Figure 7E,G). These findings are in line with the in vitro results showing that BDNF acts as a primer of platelet activation and secretion.
Discussion
In the present study, we have shown that (1) at low concentrations, BDNF primes platelets to subthreshold concentrations of classical agonists; (2) at high concentrations, BDNF induces complete, biphasic and irreversible platelet aggregation in washed human platelets; (3) platelets express a low abundance protein that is in all likelihood a truncated form of the TrkB receptor; (4) BDNF-induced platelet aggregation engages the Rho GTPase Rac-1 and downstream signaling through PKC and the PI3K pathway; (5) in response to BDNF, platelets release a broad spectrum of inflammatory and angiogenic cytokines; and (6) platelet responses are impaired at low agonist concentrations in carriers of the N187S and M189T BDNF gene variants. Taken together, these findings suggest a paracrine/autocrine role of BDNF on platelet function, and possibly other cell types, at or near a vascular lesion.
TrkB expression in platelets
Our finding that inhibition of secondary mediators of platelet aggregation with aspirin and an ADP-P2Y12 receptor antagonist prevented the secondary phase of platelet aggregation, but maintained the primary phase evoked by BDNF, suggested the presence of a specific receptor. We are not the first to investigate the presence of TrkB on platelets and platelet precursors, megakaryocytes. Two previous publications failed to detect the presence of TrkB on human platelets; however, data were not shown to support their findings.14,36 In animal studies, TrkB was expressed at very low levels in healthy rat platelets but was upregulated in platelets from tumor-bearing rats.37 The presence of TrkB on megakaryocytes is also controversial. Labouyrie et al reported high levels of expression of the truncated TrkB receptor in human bone marrow megakaryocytes, whereas Tamura et al failed to detect the TrkB receptor in a megakaryocytic cell line (however, data were not shown).38,39 Our results in human platelets from healthy volunteers suggest a truncated form of TrkB to be present, admittedly at low abundance, both at the surface and in the intracellular compartment. Arguably, the presence of the protein by mass spectrometry would have yielded a more definitive answer. The absence of TrkB in most published platelet proteomic datasets gives us pause. Because mass spectrometry is less efficient for membrane-bound proteins, low-abundance proteins, and high-molecular-weight proteins, it is possible that TrkB was simply missed due to its low levels of expression and membrane distribution.40 A single study has so far reported TrkB in human platelets in an ubiquitin-enriched platelet proteome.41 Further specialized proteomic approaches will be needed to definitely confirm the presence of TrkB in human platelets.
Signaling pathways downstream of BDNF activation in platelets
Similarly to the signaling cascade activated by BDNF in glial cells, we have found Rac1 to be important in platelets.4,42 The activation of the PKC and PI3K/Akt pathways, which triggers STAT3 activation, is an important pathway in the central nervous system promoting dendritic arborization in neurons, astrocyte survival, and glial cell differentiation.43,44 As a transcription factor, STAT3 activation by neurotrophins is often correlated with neuron survival and transcription of neuroprotective genes.45 In megakaryocytes, STAT3 has been shown to play an important role in megakaryocyte maturation and differentiation, as well as in platelet production.46 Furthermore, nongenomic roles of STAT3 have emerged in recent years, suggesting a crosstalk between STAT3 and proinflammatory cytokines in platelet signaling.47 Our finding that BDNF induces secretion of angiogenic and inflammatory cytokines in platelets warrants more detailed investigation. A limitation to this section of the study would be the use of the PKC inhibitor, BIM-1, which is known to inhibit GSK3 at nanomolar concentrations.48 Indeed, BDNF has been shown to affect GSK3β in the nervous system via the PI3K/Akt pathway.49 Further studies should be undertaken to delineate the possible roles of the PKC family and GSK3β in the ability of the truncated TrkB receptor to induce platelet activation in response to BDNF binding.
Physiological role of BDNF in platelet function
Platelets have long been recognized as reservoirs of BDNF, releasing large amounts of the neurotrophin in circulation upon activation.14 Our findings of BDNF binding to and activating platelets raise the complex question of the importance and physiological role of BDNF in platelet function. The large evidence base for BDNF involvement in neurological disorders, including ASDs, provided a strong basis for looking into platelet defects in well-characterized families.32 In the Leuven Autism Research cohort, we identified 2 families with rare variants in the BDNF gene, both in loop III of the mature protein, believed to be important in binding to the TrkB receptor.33,34 Our results indicate that the variants do not prevent the protein from being formed, as BDNF was detectable both in plasma and in platelet lysates of these participants (with the caveat that all carriers were heterozygous). However, the platelet function phenotype was indicative of a mild platelet function defect, with impaired responses to low concentrations of collagen, variable responses to ADP, and consistently lower ATP secretion in participants carrying the BDNF variants N187S and M189T. It should be noted, however, that we did not confirm whether the BDNF variants do indeed bind differently to TrkB. While protein modeling suggested the variants are likely to be pathogenic, more in-depth characterization of the mutated BDNF protein and its interaction with TrkB is needed. Notwithstanding, the phenotype seen in these 2 families is in line with our in vitro findings, where incubation of platelets with BDNF primed platelets to subthreshold concentrations of agonists, and incubation with the BDNF-neutralizing antibody prolonged collagen lag time and altered platelet response to ADP, suggesting BDNF may modulate hemostatic and thrombotic responses to vascular lesions.
These observations bear a striking resemblance with another neuropeptide shown to have similar autocrine/paracrine roles to BDNF in the nervous and vascular systems.50 Tachykinins are stored and released from platelets upon platelet activation, and they bind their cognate receptors to induce platelet aggregation that relies on secondary mediators.51 Inhibition of the tachykinin receptor has been shown to reduce thrombus formation, reduce propensity to thromboembolism, and increase bleeding time, suggesting a physiological importance in hemostasis and thrombosis.51 This striking similarity with our BDNF findings adds to the growing literature linking platelet and cognitive function,52 reinforcing the need for in-depth investigation of neuroactive substances in the vascular system.
Platelet-secreted BDNF as a paracrine regulator in the cardiovascular system
TrkB expression is not confined to the central nervous system, as the receptor has been found in many tissues such as the lungs, heart, and vascular endothelium.11,53 Increasingly, BDNF is seen as an important player in the cardiovascular system. Through activation of the truncated TrkB receptor, BDNF regulates heart contraction and long-term homeostasis in cardiomyocytes.54 BDNF also promotes angiogenesis by recruitment of endothelial cells and mobilization of hematopoietic progenitors.10,55 BDNF may protect against cardiac dysfunction and promote cell regeneration during ischemic injuries and myocardial infarction.12,56 As the largest peripheral reservoir of BDNF, platelets appear to be crucial vectors of BDNF in the cardiovascular system and active regulators of its bioavailability.
In conclusion, we have found that platelets respond to BDNF via activation of a truncated form of the TrkB receptor, engaging the Rho GTPase Rac1, signaling through the PKC and PI3K/Akt pathways, and promoting secretion of angiogenic and inflammatory cytokines. The importance of platelet-borne BDNF and its interactions with other TrkB+ cells in the vasculature, both in healthy vessels and in ischemic lesions, opens up new avenues of research into platelets as dynamic vectors of neurotrophic factors.
Acknowledgments
The authors would like to thank Louis Villeneuve at the Montreal Heart Institute core imaging facility for support with confocal microscopy and H. Uri Saragovi, Yahye Merhi, and Rahma Boulahya for their insights and suggestions. The visual abstract was created using BioRender.
This work was supported by the Canadian Institutes of Health Research (PJT-159569) and the Canada Foundation for Innovation Leaders Opportunity Fund (32797). I.B. was supported by scholarships from the Faculté de pharmacie and the Faculté des études supérieures et postdoctorales of the Université de Montréal. S.F. was supported by scholarships from the Faculté de pharmacie and the Faculté des études supérieures et postdoctorales of the Université de Montréal and the Montreal Heart Institute Foundation and is a Canadian Vascular Network Scholar. J.L.B. was supported by summer internships from the Faculté de pharmacie of the Université de Montréal. K.F. is supported by the Research Council of the University of Leuven (BOF KU Leuven, Belgium, C14/19/096). M.L. was supported by the Fonds de recherche du Québec en Santé Junior 1 Research Scholarship (33048) and is a Canada Research Chair in Platelets as biomarkers and vectors (950-232706).
Authorship
Contribution: I.B. designed the research, performed assays, collected data, analyzed and interpreted data, and wrote the manuscript; S.F., M.W., J.L.B., C.T., and M.G.B. performed assays and collected data, analyzed and interpreted data, and critically revised the manuscript; B.G.A., K.F., and T.W. analyzed and interpreted data and critically revised the manuscript; and M.L. has overseen the research group, designed the research, obtained funding, analyzed and interpreted data, and critically revised the manuscript.
Conflict-of-interest disclosure: K.F. has received unrestricted grants from CSL Behring, Novo Nordisk, Bayer, and SOBI. T.W. is a shareholder of GRAIL. M.L. has received speaker honoraria from Bayer; has received research grants to the institution from Idorsia; has served on a national advisory board for Servier; and has received in-kind and financial support for investigator-initiated grants from Leo Pharma, Roche Diagnostics, Aggredyne, and Fujimori Kogyo. The remaining authors declare no competing financial interests.
Correspondence: Marie Lordkipanidzé, Research Center, Montreal Heart Institute, 5000 rue Bélanger, Montréal, QC H1T 1C8, Canada; e-mail: marie.lordkipanidze@umontreal.ca.