

Key Points
The immunometabolite itaconate is taken up by erythroid precursors and converted to itaconyl-CoA by the CoA transferase SUGCT.
Itaconyl-CoA is a competitive inhibitor of ALAS2 and inhibits erythropoietic heme synthesis.

Abstract
As part of the inflammatory response by macrophages, Irg1 is induced, resulting in millimolar quantities of itaconate being produced. This immunometabolite remodels the macrophage metabolome and acts as an antimicrobial agent when excreted. Itaconate is not synthesized within the erythron but instead may be acquired from central macrophages within the erythroid island. Previously, we reported that itaconate inhibits hemoglobinization of developing erythroid cells. Herein we show that this action is accomplished by inhibition of tetrapyrrole synthesis. In differentiating erythroid precursors, cellular heme and protoporphyrin IX synthesis are reduced by itaconate at an early step in the pathway. In addition, itaconate causes global alterations in cellular metabolite pools, resulting in elevated levels of succinate, 2-hydroxyglutarate, pyruvate, glyoxylate, and intermediates of glycolytic shunts. Itaconate taken up by the developing erythron can be converted to itaconyl–coenzyme A (CoA) by the enzyme succinyl-CoA:glutarate-CoA transferase. Propionyl-CoA, propionyl-carnitine, methylmalonic acid, heptadecanoic acid, and nonanoic acid, as well as the aliphatic amino acids threonine, valine, methionine, and isoleucine, are increased, likely due to the impact of endogenous itaconyl-CoA synthesis. We further show that itaconyl-CoA is a competitive inhibitor of the erythroid-specific 5-aminolevulinate synthase (ALAS2), the first and rate-limiting step in heme synthesis. These findings strongly support our hypothesis that the inhibition of heme synthesis observed in chronic inflammation is mediated not only by iron limitation but also by limitation of tetrapyrrole synthesis at the point of ALAS2 catalysis by itaconate. Thus, we propose that macrophage-derived itaconate promotes anemia during an inflammatory response in the erythroid compartment.
Introduction
Induction of heme biosynthesis is a critical process in differentiating erythroid cell precursors and is ultimately responsible for the production of >400 billion hemoglobin molecules per second in adult human bone marrow.1 In metazoans, heme production in the erythroid compartment is a committed pathway that consists of 8 enzymatic steps. Erythroid heme synthesis begins with production of 5-aminolevulinic acid (ALA) via the rate-limiting enzyme ALA synthase 2 (ALAS2) and concludes with the insertion of ferrous iron into protoporphyrin IX (PPIX) by ferrochelatase (FECH)2,3 (Figure 1). ALAS2 is upregulated by erythroid-specific factors such as GATA binding factor 1 (GATA1), nuclear factor, erythroid 2 (NFE2), and erythroid Krüppel-like factor (EKLF)4,5 in the proerythroblast and basophilic stages of development in mice and humans, respectively,6 and it subsequently governs the remaining steps of erythroid differentiation.7-9 Notably, ALAS2 induction occurs after intimate contacts between a red cell progenitor and central macrophage (CM) are established in an erythroblastic island.10-12 The CM assists in erythroblast development by inhibiting apoptosis,13 facilitating iron delivery,14-16 and enhancing enucleation by pyrenocytolysis.12,17

Heme biosynthesis and the erythroblastic island. The terminal steps of erythroid development occur while erythroblasts are closely associated with a CM via αβ integrins, VCAM1, erythroblast macrophage protein (EMP), and ICAM4. Heme synthesis in this context is fully activated by the basophilic erythroblast (BasoEB) stage of development. CD163 and CD169 are macrophage-specific cell surface markers. Heme synthesis enzymes are in bold green text. CFU-E, colony forming unit–erythroid; CPOX, coproporphyrinogen III oxidase; HMBS, hydroxymethylbilane synthase; HSC, hematopoietic stem cell; OrthoEB, orthochromatic erythroblast; PBGS, porphobilinogen synthase; PolyEB, polychromatic erythroblast; PPOX, protoporphyrinogen oxidase; ProEB, proerythroblasts; RBC, red blood cell; SCoA, succinyl-CoA; UROD, URO decarboxylase; UROS, uroporphyrinogen III synthase.
Heme biosynthesis and the erythroblastic island. The terminal steps of erythroid development occur while erythroblasts are closely associated with a CM via αβ integrins, VCAM1, erythroblast macrophage protein (EMP), and ICAM4. Heme synthesis in this context is fully activated by the basophilic erythroblast (BasoEB) stage of development. CD163 and CD169 are macrophage-specific cell surface markers. Heme synthesis enzymes are in bold green text. CFU-E, colony forming unit–erythroid; CPOX, coproporphyrinogen III oxidase; HMBS, hydroxymethylbilane synthase; HSC, hematopoietic stem cell; OrthoEB, orthochromatic erythroblast; PBGS, porphobilinogen synthase; PolyEB, polychromatic erythroblast; PPOX, protoporphyrinogen oxidase; ProEB, proerythroblasts; RBC, red blood cell; SCoA, succinyl-CoA; UROD, URO decarboxylase; UROS, uroporphyrinogen III synthase.
The inability to produce sufficient numbers of normal erythrocytes results in anemia. Although anemia is the most prevalent of all blood disorders, it is generally underappreciated and undertreated despite its impact on quality of life and the economy.18 Iron-deficiency anemia (IDA), followed in frequency by anemia of inflammation (AI), are major contributors to the morbidity and mortality of chronic illnesses.19 IDA is characterized as a hypochromic, microcytic anemia, whereas AI is usually a normochromic, normocytic anemia.20 Microcytosis and loss of ALAS2 activity have been observed in the bone marrow of patients with rheumatoid arthritis (RA).21 It has been postulated that systemic hypoferremia common to AI is causative. However, ferrokinetic studies in patients with RA do not indicate reduced iron supply to bone marrow,22 and FECH activity in patients with RA is normal.21 This finding contrasts with what is found in IDA,23 in mouse models of IDA in which IRP2 is knocked out with decreased transferrin receptor expression,24 and in low cellular iron conditions in which FECH activity is proposed to be limited by decreased iron-sulfur cofactor assembly.25 In samples of whole bone marrow from a patient with RA, ALA synthesis is reduced to a far greater magnitude than in purified erythroblasts,21 suggesting that resident immunoactivated leukocytes contribute to the loss of ALAS2 activity and corresponding anemia. Together, these observations strongly suggest that factors other than the availability of iron for erythropoiesis must be involved to limit heme synthesis in AI.
We hypothesize that alterations in the erythropoietic program during inflammation are mediated by the CM in erythroid islands. Lipopolysaccharide (LPS)-mediated inflammation in mice results in anemia via inhibition of erythropoiesis in bone marrow at an early stage, with an observed depletion of proerythroblasts and basophilic erythroblasts,26 the stages during which ALAS2 is first induced.6 A variety of factors are produced and excreted by macrophages as part of their inflammatory response. In the last decade, multiple studies have revealed that macrophages synthesize itaconate (supplemental Figure 1A), an α,β-unsaturated carboxylic acid and close analogue of succinate (methylenesuccinic acid) (supplemental Figure 1B), at the outset of an inflammatory response.27-32 Itaconate is generated in millimolar quantities from the TCA cycle intermediate cis-aconitate via immune-response gene 1 (IRG1) protein, which can be induced by proinflammatory factors such as LPS and interferon-γ in bone marrow–derived macrophages (BMDMs).29,31 Importantly, itaconate is secreted by macrophages,28,30,32 including BMDMs, and has recently been identified in local tissues during sepsis30 and in the plasma of patients with RA.33 Thus, it is believed that itaconate has characteristics that affect metabolism via autocrine, paracrine, and endocrine processes.34 Itaconate inhibits succinate dehydrogenase (SDH)31,32,35 and has been associated with succinate-driven stabilization of hypoxia-inducible factor-1α and proinflammatory interleukin 1β (IL1β) production.36 It also attenuates liver phosphofructokinase 2 (PFK2) activity in glycolysis37 and further disrupts the TCA cycle by blocking substrate-level phosphorylation.38 Older publications proposed that itaconate is converted to itaconyl-CoA (supplemental Figure 1C) by reverse succinyl CoA synthetase (SCS) activity with adenosine triphosphate (ATP) or guanosine triphosphate (GTP) supplementation,39,40 although direct evidence for this theory was lacking. Itaconate has been shown to have antimicrobial activity by inhibition of the glyoxylate shunt41-43 and as itaconyl-CoA by inactivation of vitamin B12.44,45 B12 can be scavenged by microbes in their host organisms,46 and the suicide inhibition of the B12-dependent methylmalonic acid mutase (MUT) by itaconyl-CoA leads to the accumulation of toxic propionyl-CoA and may curtail cholesterol-dependent growth of Mycobacterium tuberculosis.45
Herein we report that the immunometabolite itaconate taken up by the developing erythron is converted to itaconyl-CoA and controls porphyrin synthesis at the point of ALAS2 catalysis. We also show global metabolic changes that occur in developing erythrocytes upon itaconate treatment suggest alterations in carbon flux for heme synthesis. Moreover, we provide evidence that itaconyl-CoA is produced by a CoA transferase and not SCS in red cell progenitors. We believe this mechanism as a whole is crucial during acute and/or chronic conditions of anemia when itaconate has been shown to accumulate.
Methods
Reagents
All reagents were obtained from Sigma unless otherwise noted. 13C5-itaconate was synthesized by the Metabolite Standards Synthesis Core at SRI International, arranged through the National Institutes of Health Common Funds Metabolomics initiative.47 Details regarding the preparation of itaconyl-CoA are provided in the supplemental Methods.
Cell culture
DS19 murine erythroleukemia (MEL) cells48,49 were used as erythroblast models and propagated in Dulbecco’s modified Eagle medium/F12 media (Corning) containing 25 mM glucose and 1 mM sodium pyruvate supplemented with 2 mM glutamine, 10% fetal bovine serum, and 1% penicillin-streptomycin under a humidified 5% carbon dioxide atmosphere. Details of the culture methods are presented in the supplemental Methods.
Heme and porphyrin analysis
MEL cells treated with or without 2.5 mM unlabeled itaconate and 1.5% dimethyl sulfoxide were analyzed in quadruplicate (n = 4 biological replicates) for heme, PPIX, and pathway intermediates 8-, 7-, 6-, 5-, and 4-carboxylporphyrins (for isomers I and III) by using high-performance liquid chromatography, as reported previously.50 Details are presented in the supplemental Methods.
Expression and purification of recombinant enzymes
The design and preparation of expression plasmids, recombinant protein expression in Escherichiacoli, and purification are detailed in the supplemental Methods.
Spectrophotometric assays and kinetics
ALAS activity assays were conducted either in coupled assays or with derivatized ALA detection via Ehrlich’s modified reagent, with microscale modifications as detailed in the supplemental Methods. For inhibition studies, inhibitory constant and mode of inhibition were determined by titrating 10 to 200 μM succinyl-CoA in the presence of 50 mM glycine substrate and 0, 200, and 1000 μM itaconyl-CoA. Michaelis-Menten nonlinear regression analysis was subsequently conducted on data for 3 separate protein preparations (n = 3 biological replicates) with Prism software (GraphPad 8.4).
Pig heart SCS-G (Sigma) and recombinant SCS-A were assayed for reverse activity (GTP or ATP consumption as appropriate) with succinate and/or itaconate as substrate(s) using a direct spectrophotometric assay for CoA ester bond formation at 225 to 235 nm51 as detailed in the supplemental Methods.
High-performance liquid chromatography assays of CoA transferase activity
SUGCT (succinyl-CoA:glutarate-CoA transferase) activity was determined as reported in the literature,52 with minor changes as detailed in the supplemental Methods.
Results
Itaconate decreases heme and porphyrin synthesis in erythroid precursors
Previously, it was shown that when medium from RAW264.7 macrophages treated with LPS was added to differentiating MEL cells, the level of heme production decreased.53 We have verified these experimental results knowing that these cells produce and excrete itaconate under these conditions (supplemental Figure 2). Previously, we established that the inflammatory metabolite itaconate attenuates hemoglobin production in MEL cells undergoing erythroid differentiation and that this block is bypassed by providing the cells with ALA.54 Here we show that heme, PPIX, and all carboxyl porphyrin intermediates are significantly decreased during MEL cell differentiation in the presence of itaconate (Figure 2A-D). These data indicate that inhibition of heme synthesis occurs upstream of uroporphyrinogen decarboxylase (Figure 1). To better target the point of inhibition, we examined the effects of itaconate on transgenic MEL cells overexpressing hsALAS2. Normally, these cells overproduce porphyrins because pathway regulation is bypassed by the expression of excess ALAS2. Treatment of these cells with itaconate reduces porphyrin-induced fluorescence (Figure 2E), suggesting that blockage of the pathway occurs before tetrapyrrole formation.
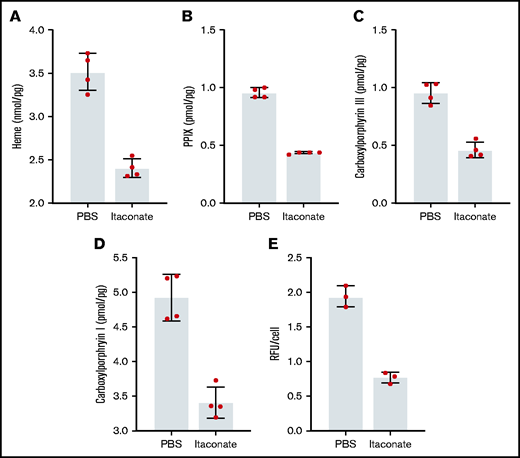
Porphyrin and heme analysis of MEL cells treated with itaconate. Heme (A), PPIX (B), and total carboxyl porphyrins (C,D) were determined by high-performance liquid chromatography for wild-type MEL cells differentiated in dimethyl sulfoxide media for 72 hours. (E) Relative fluorescence of cyclic porphyrins in media from MEL cell cultures overexpressing Homo sapiens ALAS2 (hsALAS2) over 72 hours without dimethyl sulfoxide induction. Itaconate concentrations were 2.5 mM in all experiments shown here. Bars represent means ± 1 standard deviation (n = 3-4 biological replicates as indicated red dots). Multiple Student t tests combined with Bonferroni analysis produced P values <.01 for all phosphate-buffered saline (PBS) vs itaconate comparisons shown. RFU, relative fluorescence units.
Porphyrin and heme analysis of MEL cells treated with itaconate. Heme (A), PPIX (B), and total carboxyl porphyrins (C,D) were determined by high-performance liquid chromatography for wild-type MEL cells differentiated in dimethyl sulfoxide media for 72 hours. (E) Relative fluorescence of cyclic porphyrins in media from MEL cell cultures overexpressing Homo sapiens ALAS2 (hsALAS2) over 72 hours without dimethyl sulfoxide induction. Itaconate concentrations were 2.5 mM in all experiments shown here. Bars represent means ± 1 standard deviation (n = 3-4 biological replicates as indicated red dots). Multiple Student t tests combined with Bonferroni analysis produced P values <.01 for all phosphate-buffered saline (PBS) vs itaconate comparisons shown. RFU, relative fluorescence units.
In agreement with our MEL cell data,54 hemoglobin production in EPO-induced human TF1 cells is diminished with itaconate and can be rescued by ALA (supplemental Figure 3A). However, we also noted a significant increase in cell proliferation with itaconate. Unlike MEL cells55 or erythroid-differentiated CD34+ cells,56 TF1 cells express the succinate cognate G protein–coupled receptor 91 (GPR91, or SUCNR1).57 Because SUCNR1 is sensitive to itaconate58 and known to stimulate ERK signaling, we added combinations of itaconate and the SUCNR1 antagonist compound 4c (gift of Xin Li) or MEK inhibitor U0126 to differentiating TF1 cultures to assess the impact of the SUCNR1-MAPK/ERK pathway on differentiation. The inhibitors independently rescued hemoglobin levels and proliferation rates measured in control cells, suggesting that in TF-1 cells, the observed decrease in hemoglobinization is at least partially due to nonspecific itaconate binding to SUCNR1 and the associated increase in TF1 cell proliferation via the SUCNR1-ERK signaling axis57 (supplemental Figure 3B-C).
Itaconate is imported by erythroid precursors and converted to itaconyl-CoA
Although it is known that itaconate is actively secreted by macrophages and that itaconate added to cultures of MEL cells results in decreased hemoglobinization, it was necessary to show that itaconate can be imported by developing erythroid cells. Mass spectrometry–based metabolomic tracing of dimethyl sulfoxide–induced MEL cells cultured with uniformly labeled (13C5)-itaconate exhibited the intracellular accumulation of the same species (Figure 3A). In support of this observation, mitochondrial itaconate transporters, including SLC25A10 (succinate) and SLC25A1 (citrate), have been identified in vitro,59 making movement of exogenously supplied itaconate into the mitochondrial compartment feasible. These transporters are expressed at significant levels in erythroid-differentiating MEL55 and CD34+ cells.56 With respect to plasmalemmal counterpart(s), a cell surface itaconate transporter has not yet been identified in any mammalian cell type, although our data clearly show that a mechanism for itaconate uptake by cells must exist.60

Liquid chromatography–mass spectrometry analysis of differentiating MEL cells treated with labeled itaconate. MEL cultures were induced with dimethyl sulfoxide and treated with phosphate-buffered saline (PBS) or 1 mM labeled itaconate for 72 hours. Select metabolites targeted by liquid chromatography–mass spectrometry analysis are given on the y-axes. Analyses were conducted as described in the Methods section. Bars represent means ± 1 standard deviation (n = 4 biological replicates). Multiple Student t tests combined with Bonferroni analysis produced P values <.01 for all PBS vs 13C5-itaconate for the metabolites (A) 13C5-itaconate, (B) 13C5-itaconyl-CoA, (D) succinyl-CoA, (E) propionyl-CoA, (F) propionyl-carnitine, and (G) inositol, comparisons except (C) succinate (0.024) and (H) AMP (0.034).
Liquid chromatography–mass spectrometry analysis of differentiating MEL cells treated with labeled itaconate. MEL cultures were induced with dimethyl sulfoxide and treated with phosphate-buffered saline (PBS) or 1 mM labeled itaconate for 72 hours. Select metabolites targeted by liquid chromatography–mass spectrometry analysis are given on the y-axes. Analyses were conducted as described in the Methods section. Bars represent means ± 1 standard deviation (n = 4 biological replicates). Multiple Student t tests combined with Bonferroni analysis produced P values <.01 for all PBS vs 13C5-itaconate for the metabolites (A) 13C5-itaconate, (B) 13C5-itaconyl-CoA, (D) succinyl-CoA, (E) propionyl-CoA, (F) propionyl-carnitine, and (G) inositol, comparisons except (C) succinate (0.024) and (H) AMP (0.034).
Further tracing analysis of the differentiating MEL cell samples indicates that one metabolic fate of internalized 13C5-itaconate is 13C5-itaconyl-CoA (Figure 3B). This result was unsurprising based on 50-year-old data suggesting that mammalian mitochondrial SCS may use itaconate as an alternative substrate for succinate in ATP- or GTP-driven synthesis of itaconyl-CoA.39,40
Itaconate affects flux through the erythroblast TCA cycle and glycolytic shunts
Liquid chromatography–mass spectrometry analysis of differentiating MEL cells treated with 1 mM 13C5-itaconate or 2.5 mM unlabeled itaconate showed significant differences in TCA cycle metabolite levels from cells differentiated in the absence of itaconate. Specifically, succinate (Figure 3C; supplemental Figures 4, 5A, and 6) and succinyl-CoA (Figure 3D) were elevated in itaconate-treated cultures, although neither was labeled by 13C5-itaconate. The finding of elevated succinate levels was not surprising given the established direct inhibitory action of itaconate on SDH in other cell types.31,32 Fumarate increased as well with itaconate (supplemental Figure 4A) despite putative SDH inhibition. The α-ketoglutarate derivative 2-hydroxyglutarate (supplemental Figures 4A, 5A, and 6) also accumulated, and increased glutamic acid was found in the media (supplemental Figure 6B). Both these observations may be due to product inhibition of α-ketoglutarate dehydrogenase caused by the elevated levels of succinyl-CoA.61
Our results also suggest that glycolysis is disrupted by itaconate during terminal erythropoiesis. Intermediates of glycolytic shunts such as the pentose phosphate and serine-glycine synthesis pathways increased compared with controls, including AMP (Figure 3E), adenosine and thymine (supplemental Figure 4A), serine (supplemental Figures 4, 5, and 6), and glycine (supplemental Figure 6A). Unexpectedly, levels of glutathione in the culture media were also elevated (supplemental Figures 4B and 6B), as itaconate has been reported to directly alkylate glutathione in LPS-activated macrophages.62 Myoinositol (supplemental Figures 4A, 5A, and 6A) and inositol (Figure 3F), products of glucose-6-phosphate metabolism, increased with itaconate as well. Allosteric inhibition of the glycolysis-activating PFK2 by itaconate, a phosphoenolpyruvate analogue, has been previously described in the liver37 and could explain these observations in our differentiating erythroid progenitors.
Itaconate impairs metabolism of amino acids and fatty acid oxidation compounds
It is well established that aliphatic amino acids and odd-chain fatty acids are ultimately catabolized to propionyl-CoA and then methylmalonyl-CoA by propionyl-CoA carboxylase and B12-dependent methylmalonyl-CoA mutase (MUT), respectively. Further metabolomic data gathered here indicate that flux through these pathways is attenuated in differentiating erythroblast cultures treated with itaconate, likely via itaconyl-CoA and its ability to inactivate B12 and MUT activity as previously discussed in adipocytes44 and macrophages.45 Namely, we found increased levels of propionyl-CoA (Figure 3G), propionyl-carnitine (Figure 3H), and methylmalonic acid (supplemental Figures 4, 5, and 6) with itaconate treatment. Upstream of propionyl-CoA, we measured elevations in the odd-chain fatty acids heptadecanoic acid (supplemental Figures 4A and 6A) and nonanoic acid (supplemental Figure 6B), as well as the aliphatic amino acids threonine, valine, methionine, and isoleucine (supplemental Figure 6). Notably, itaconyl-CoA accumulations may also explain increased pyruvate and glyoxylate levels (supplemental Figures 4B, 5B, and 6B). Itaconyl-CoA is an alternative substrate of methylglutaconase and is converted by this enzyme to citramalyl-CoA.40,44 It is therefore reasonable to believe that citramalyl-CoA lyase (CLYBL) catalyzes the cleavage of citramalyl-CoA to pyruvate and acetyl-CoA40 in lieu of metabolizing glyoxylate.63
Itaconyl-CoA is produced from itaconate via SUGCT but not SCS
Indirect evidence from approximately one-half century ago has been the basis for the proposal that itaconate is converted to itaconyl-CoA by SCS,39,40 and in the absence of definitive data, it has been assumed by others to occur in this fashion.38,64 However, we observed that neither ATP- nor GTP-specific SCS is able to use itaconate under the same conditions as succinate in vitro (Figure 4A-B; supplemental Figure 7A-B). Furthermore, we observed that SCS activity is inhibited by itaconate in a dose-dependent manner (Figure 4C).
![Inhibition of the reverse SCS reaction by itaconate. Succinate but not itaconate at 10 mM concentrations is metabolized by pig heart SCS-GTP (SCS-G) (A) and recombinant SCS-ATP (SCS-A) (B). (C) Itaconate inhibits SCS-A at the indicated ratios using 50 mM succinate in all reactions. Summary bar plots represent mean rates of acyl-CoA formation at 225 to 235 nm. The corresponding UV scans for panels A and B are provided in supplemental Figure 7. Bars represent means and error bars = 1 standard deviation (n = 2-3 biological replicates). Panel A: Student t test P value = .0001. Panel C: one-way analysis of variance P value = .0022 with Bonferroni post hoc test P values = .026, .0022, and .15 (not significant [ns]) for control vs 1:200, control vs 1:50, and 1:200 vs 1:50, respectively.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/23/10.1182_bloodadvances.2021004750/2/m_advancesadv2021004750f4.png?Expires=1763680085&Signature=Ujkh6wHiHF3231L~C5TanI2NURM0TQfhLTvR66AvkcH~rhH0jGkYVEFWAn~9xSJQgccxXTCGijS50Y-lgsrnHM~0QsHOxG1ogo~q7IvCfqlhOsSUVqjv6eA6lfcDISAQ6cx-9iCKXRXGUSLLv2Ivd9VX8muBNIIRD0EEaAkLkQ0dA8hRtpX32VaYvQvDmvu4qXwIWBzXoG~zd2NfFPysguJeciJMuJlyZYVuP2XgRIhKDIvev4cpCxYd-b96m9L2isZ2JfJUvjhL3SJMnRJN77F0NKdhxpf~YDVXUMvGs0WdWdHrvvia80aZZdNgSUy8dTjVTXcBVmLGFO-KLtXPuw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Inhibition of the reverse SCS reaction by itaconate. Succinate but not itaconate at 10 mM concentrations is metabolized by pig heart SCS-GTP (SCS-G) (A) and recombinant SCS-ATP (SCS-A) (B). (C) Itaconate inhibits SCS-A at the indicated ratios using 50 mM succinate in all reactions. Summary bar plots represent mean rates of acyl-CoA formation at 225 to 235 nm. The corresponding UV scans for panels A and B are provided in supplemental Figure 7. Bars represent means and error bars = 1 standard deviation (n = 2-3 biological replicates). Panel A: Student t test P value = .0001. Panel C: one-way analysis of variance P value = .0022 with Bonferroni post hoc test P values = .026, .0022, and .15 (not significant [ns]) for control vs 1:200, control vs 1:50, and 1:200 vs 1:50, respectively.
Inhibition of the reverse SCS reaction by itaconate. Succinate but not itaconate at 10 mM concentrations is metabolized by pig heart SCS-GTP (SCS-G) (A) and recombinant SCS-ATP (SCS-A) (B). (C) Itaconate inhibits SCS-A at the indicated ratios using 50 mM succinate in all reactions. Summary bar plots represent mean rates of acyl-CoA formation at 225 to 235 nm. The corresponding UV scans for panels A and B are provided in supplemental Figure 7. Bars represent means and error bars = 1 standard deviation (n = 2-3 biological replicates). Panel A: Student t test P value = .0001. Panel C: one-way analysis of variance P value = .0022 with Bonferroni post hoc test P values = .026, .0022, and .15 (not significant [ns]) for control vs 1:200, control vs 1:50, and 1:200 vs 1:50, respectively.
In an effort to identify a mammalian enzyme for itaconyl-CoA production, we first turned to bacterial enzymes capable of itaconyl-CoA synthesis. It is known that Pseudomonas aeruginosa itaconyl-CoA transferase (PaIct) is an enzyme capable of forming itaconyl-CoA from succinyl-CoA and itaconate without added nucleotides.64 Using a bioinformatics approach, we found that mammalian SUGCT (encoded by the C7ORF10 gene) is a homologue of the P aeruginosa PaIct (Figure 5A). Others have reported SUGCT activity with the canonical substrate glutaryl-CoA.52 We cloned, expressed, and purified SUGCT and determined that it effectively catalyzes the production of itaconyl-CoA using succinyl-CoA (Figure 5B). In agreement with transcript data in MEL cells,55 immunoblotting showed that SUGCT protein is present in mitochondria isolated from induced and uninduced MEL cells (Figure 5C). Thus, SUGCT is likely responsible for itaconyl-CoA synthesis in developing erythrocytes.

SUGCT converts itaconate to itaconyl-CoA. (A) Human SUGCT is homologous to the P aeruginosa itaconyl-CoA transferase (PaIct). Black and gray boxes highlight identical residues and conservative substitutions, respectively. (B) Discontinuous high-performance liquid chromatography (HPLC) assays showed that SUGCT generates itaconyl-CoA from succinyl-CoA and itaconate in vitro. (C) Immunoblotting of MEL mitochondria from cells treated with and without 2.5 mM itaconate and dimethyl sulfoxide (DMSO) for 72 hours. MEL mitochondrial lysates were probed with anti-ALAS2, anti-FECH, and anti-SUGCT primary antibodies. HPLC assays were performed on a C18 reverse-phase column with UV detection (mAU = milli-absorbance units at 254 nm). Optimal autoexposure within the linear dynamic range was performed for each immunoblot on the ChemiDoc imaging system (Bio-Rad). The HPLC chromatogram (B) and western blot (C) shown are representative of n = 2 biological replicates.
SUGCT converts itaconate to itaconyl-CoA. (A) Human SUGCT is homologous to the P aeruginosa itaconyl-CoA transferase (PaIct). Black and gray boxes highlight identical residues and conservative substitutions, respectively. (B) Discontinuous high-performance liquid chromatography (HPLC) assays showed that SUGCT generates itaconyl-CoA from succinyl-CoA and itaconate in vitro. (C) Immunoblotting of MEL mitochondria from cells treated with and without 2.5 mM itaconate and dimethyl sulfoxide (DMSO) for 72 hours. MEL mitochondrial lysates were probed with anti-ALAS2, anti-FECH, and anti-SUGCT primary antibodies. HPLC assays were performed on a C18 reverse-phase column with UV detection (mAU = milli-absorbance units at 254 nm). Optimal autoexposure within the linear dynamic range was performed for each immunoblot on the ChemiDoc imaging system (Bio-Rad). The HPLC chromatogram (B) and western blot (C) shown are representative of n = 2 biological replicates.
Itaconyl-CoA inhibits, but is not a substrate of, ALAS2
Itaconyl-CoA (supplemental Figure 1C) is a close analogue of succinyl-CoA (supplemental Figure 1D) and may affect ALAS2 enzyme activity. Although itaconate does not affect ALAS2 activity (supplemental Figure 8A), discontinuous kinetic assays revealed itaconyl-CoA to be a competitive inhibitor of recombinant ALAS2 with an inhibitory constant of 100 ± 20 μM (Figure 6). We measured the Michaelis constant of succinyl-CoA to be 10 ± 2 μM for the uninhibited enzyme, which agrees with the literature for the mammalian enzyme.65-69 Thus, itaconyl-CoA could be partially responsible for the observed increase in succinyl-CoA in induced MEL cells (Figure 3D). Continuous coupled ALAS assays were also performed because ALA derivatives synthesized from other acyl-CoA substrates have shown unpredictable stability in the α-aminoketone formed via reaction with acetylacetone in the discontinuous assay protocol.66 The results of the coupled assays verify that itaconyl-CoA is not an alternative ALAS2 substrate even at millimolar concentrations (supplemental Figure 8B). This finding explains the alterations in heme precursor levels (Figure 2) and the global alterations in metabolites.
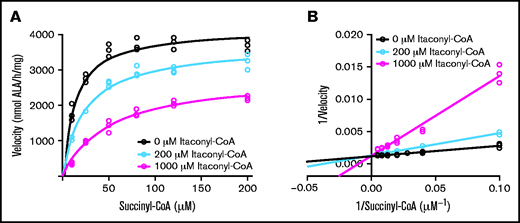
ALAS2 inhibition by itaconyl-CoA. Kinetic progress curves (A) and Lineweaver-Burk plot (B) indicating competitive inhibition of ALAS2 with an inhibitory constant = 100 ± 20 μM (mean ± 1 standard deviation) for itaconyl-CoA. Michaelis constant = 10 ± 2 μM (mean ± 1 standard deviation) for succinyl-CoA. Discontinuous colorimetric assays were conducted for n = 3 biological replicates (separate protein preparations).
ALAS2 inhibition by itaconyl-CoA. Kinetic progress curves (A) and Lineweaver-Burk plot (B) indicating competitive inhibition of ALAS2 with an inhibitory constant = 100 ± 20 μM (mean ± 1 standard deviation) for itaconyl-CoA. Michaelis constant = 10 ± 2 μM (mean ± 1 standard deviation) for succinyl-CoA. Discontinuous colorimetric assays were conducted for n = 3 biological replicates (separate protein preparations).
Discussion
Itaconate is an antimicrobial compound whose impact on mammalian liver metabolism was first explored in the 1940s,39,70,71 and we provide evidence here that itaconate has a significant impact on the erythroid compartment (Figure 7; supplemental Figure 9). Immunoactivated macrophages have been identified as the source of itaconate via IRG1 catalysis.29-32 Importantly, measurable quantities of itaconate have been found in bronchial lavages of patients with severe lung infections30 and in blood plasma of patients with RA.33 Experiments examining the impact of RA on the erythroid compartment have shown diminished ALAS2 activity in microcytic red cell precursors, most significantly in the context of whole bone marrow samples containing immunoactivated leukocytes such as BMDMs.21 Based on the results of this study, we propose that itaconate negatively alters heme biosynthesis and erythroid differentiation at the point of ALAS2 catalysis in the context of an inflammatory erythroblastic island. Specifically, we have determined that heme, PPIX, and total carboxyl porphyrin levels are decreased in MEL cells differentiated in the presence of itaconate or media from immunoactivated, itaconate-generating macrophages. Addition of ALA to itaconate-treated cultures rescues hemoglobinization, as described previously.54 13C tracing data show that itaconate is imported by differentiating MEL cells and is subsequently metabolized to itaconyl-CoA. Levels of succinyl-CoA are also increased in these cells with itaconate supplementation, suggesting that ALAS2 is directly affected. Indeed, our kinetic assays showed that itaconyl-CoA is a competitive inhibitor of ALAS2 without acting as an alternative substrate.

Model of the erythroblastic island during an inflammatory response. Itaconate is produced via IRG1 in immunoactivated blood island macrophages and excreted to the surrounding milieu. Subsequent import and metabolism of itaconate to itaconyl-CoA by proximal red cell precursors result in inhibition of ALA synthesis and cellular hemoglobinization at the point of ALAS2 catalysis. Interleukin 6 (IL6), tumor necrosis factor-α (TNFα), and interleukin 1β (IL1β) are proinflammatory cytokines. GSH, glutathione; IFNGR, interferon-γ (IFNγ) receptor; TLR4, Toll-like receptor 4.
Model of the erythroblastic island during an inflammatory response. Itaconate is produced via IRG1 in immunoactivated blood island macrophages and excreted to the surrounding milieu. Subsequent import and metabolism of itaconate to itaconyl-CoA by proximal red cell precursors result in inhibition of ALA synthesis and cellular hemoglobinization at the point of ALAS2 catalysis. Interleukin 6 (IL6), tumor necrosis factor-α (TNFα), and interleukin 1β (IL1β) are proinflammatory cytokines. GSH, glutathione; IFNGR, interferon-γ (IFNγ) receptor; TLR4, Toll-like receptor 4.
The widely accepted explanation for decreased ALAS2 activity in inflammatory states such as AI is that systemic iron is sequestered via hepcidin72 and could therefore downregulate iron-dependent ALAS2 synthesis. Mature ALAS2 messenger RNA contains a 5' iron-responsive element that is lacking in the housekeeping ALAS1 enzyme,73 and this element is bound by apo iron-regulatory proteins IRP1 and IPR2 when erythroid iron levels are low, effectively blocking ALAS2 translation.24,74 The impact of IRP1 on ALAS2 protein levels in particular has been described in cases of sideroblastic anemia caused by loss of glutaredoxin 575 and mitoferrin-1–deficient porphyria.76 However, others have reported that erythroblast iron delivery during terminal differentiation bypasses the cytosolic iron pool for IRP2, and IRP numbers in general appear to be overwhelmed late in erythroid differentiation when heme synthesis is massively upregulated.77 A more recent study has verified low levels of translated Irp1 and Irp2 relative to Alas2 at the basophilic erythroblast stage in mice and has provided evidence that heme-regulated eukaryotic initiation factor 2 α (eIF2α) kinase is the predominant regulator of hemoglobin synthesis in iron-replete and iron-deficient conditions in these cells.78 Perhaps most significantly, ferrokinetic data from RA patient samples do not indicate reduced iron supply to the marrow,22 and iron-dependent FECH activity is unaffected.21 Thus, we postulate that itaconate is a significant factor in AI by downregulating ALAS2 activity and porphyrin synthesis in the bone marrow during an inflammatory response.
Additional data also support many of the previously substantiated effects of itaconate on glycolysis,37 B12 metabolism,44,45 and the TCA cycle29,31,32 in other tissues (supplemental Figure 9). For example, we see increases in compounds such as inositol as well as products of the pentose phosphate and serine-glycine synthesis pathways, possibly due to allosteric inhibition of PFK2.37 Elevated levels of aliphatic amino acids (eg, valine), odd-chain fatty acids (eg, heptadecanoic acid), and propionyl-CoA metabolites suggest that itaconyl-CoA compromises B12 constitution in erythroblasts and irreversibly inhibits the MUT enzyme as seen in adipocytes and immunoactivated macrophages.44,45 In addition to succinyl-CoA, elevations in succinate and fumarate were observed with itaconate treatment. It is tempting to postulate that succinate accumulation is caused by itaconate-driven inhibition of SDH and accounts for succinyl-CoA production via the reverse SCS reaction. However, this does not justify the increase in fumarate. We also showed in vitro that itaconate inhibits reverse SCS activity and is not converted to itaconyl-CoA by SCS as proposed by others.39,40 Our results fit with those of a more recent study showing that itaconate impairs substrate-level phosphorylation in LPS-stimulated macrophages in respiratory distress and behaves similarly to a specific SCS inhibitor in these cells.38
Bioinformatics and immunoblotting data support the hypothesis that exogenous itaconate is derivatized to itaconyl-CoA by SUGCT in red cell progenitors. In addition, succinate contributes little to the supply of heme carbon during erythroid differentiation, and succinyl-CoA used for heme synthesis is supplied primarily through a separate pool of α-ketoglutarate dehydrogenase acting outside of the TCA cycle.54 ALAS2 inhibition thus provides a valid explanation for both increased succinyl-CoA and decreased porphyrin synthesis when erythroid precursors are exposed to itaconate. The global metabolic changes that occurred from treatment of itaconate to developing erythroid cells show a direct effect of itaconate on multiple metabolic processes, independent of additional cytokines and other immunoregulated molecules. Although some of these metabolites are clearly upstream and linked to heme synthesis in the developing erythron, others may be downstream of the process. Follow-up experiments to determine the metabolic connections and temporal response will be necessary to determine the impact of itaconate in the erythroblastic island.
Acknowledgments
The authors thank D. Bishop (Mount Sinai), A. Cantor (Boston Children’s Hospital), and X. Li (New York University) for providing some reagents used in this study.
This work was supported by National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grants DK11653 and DK110858 via Pilot and Feasibility Grants from the University of Utah Center for Iron and Heme Disorders (A.E.M.) and U54DK110858 and U24DK126127 (J.D.P.). Mass spectrometry equipment was obtained through 1S10OD016232-01 and 1S10OD021505-01 (J.E.C.).
Authorship
Contribution: J.R.M. designed experiments, analyzed data, and wrote the paper; J.E.C. and H.A.B. performed research and analyzed data; J.E.C., A.E.M., and J.D.P. analyzed data and edited the paper; and H.A.D. designed experiments, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Harry A. Dailey, Department of Microbiology, 230 Coverdell Center, University of Georgia, Athens, GA 30602; e-mail: hdailey@uga.edu.



