Key Points
Misdiagnosed primary ITP in children is rare in the international prospective Pediatric and Adult Registry on Chronic ITP.
Infectious and systemic autoimmune disorders are the most frequent causes of secondary ITP in children.

Abstract
Primary immune thrombocytopenia (ITP) in children is a diagnosis of exclusion, but cases of secondary ITP and nonimmune thrombocytopenia (non-IT) are generally difficult to recognize in a timely fashion. We describe a pediatric population with a revised diagnosis of secondary ITP or non-IT within 24 months of follow-up. Data were extracted from the Pediatric and Adult Registry on Chronic ITP, an international multicenter registry collecting data prospectively in patients with newly diagnosed primary ITP. Between 2004 and 2019, a total of 3974 children aged 3 months to 16 years were included. Secondary ITP and non-IT were reported in 113 patients (63 female subjects). Infectious (n = 53) and autoimmune (n = 42) diseases were identified as the main causes, with median ages at diagnosis of 3.2 years (interquartile range: 1.2; 6.7 years) and 12.4 years (interquartile range: 7.6; 13.7 years), respectively. Other causes included malignancies, aplastic anemia, immunodeficiency, and drug use. Patients with malignancy and aplastic anemia had significantly higher initial platelet counts (37 and 52 × 109/L) than did those with infection or autoimmune diseases (12 and 13 × 109/L). Characteristics of patients with secondary ITP due to infection were similar to those of children with primary ITP at first presentation, indicating similar mechanisms. Significant differences were found for age, sex, comorbidities, initial bleeding, sustained need for treatment, and disease persistence for the remaining noninfectious group compared with primary ITP. Based on our findings, we propose a diagnostic algorithm that may serve as a basis for further discussion and prospective trials.
Introduction
Immune thrombocytopenia (ITP) is an autoimmune disorder resulting from various etiologies that is characterized by increased platelet destruction and impaired production leading to a decrease in the platelet count. In secondary ITP, thrombocytopenia can be linked to an underlying condition, whereas no apparent cause can be found in primary ITP.1 Early discrimination between patients with primary or secondary ITP and nonimmune thrombocytopenia (non-IT; eg, bone marrow failure and congenital thrombocytopenia) is important, considering that clinical management protocols and prognoses may differ.2-4
Primary ITP in children is a diagnosis of exclusion, and no laboratory tests to confirm ITP are available. According to international practice guidelines, medical and family history-taking, clinical examination, complete blood count assessment, and blood smear analysis are sufficient to diagnose the primary form.5,6 In children, ITP is often a benign, self-limiting condition, and a watch-and-wait strategy is recommended in those experiencing no or mild bleeding.5-9 Secondary ITP and non-IT are rare and sometimes difficult to recognize in children with suspected or newly diagnosed ITP. Moreover, other manifestations of the underlying disease may emerge only during the follow-up period.10,11 Red flags that raise the suspicion of secondary ITP and other nonimmune causes of thrombocytopenia have been proposed in the last few years and include positive family history, older age (adolescence), chronic ITP, platelet size either above or below the normal range, moderate (instead of severe) thrombocytopenia at first presentation, nonresponse to first-line treatments, and new symptoms or laboratory abnormalities during the disease course.12-21 Despite growing awareness of the differential diagnosis of primary ITP, secondary ITP and non-IT seem to be frequently identified with considerable delay, and thus the diagnostic workflow may benefit from better definition and validation.
The rate of secondary ITP in newly diagnosed, persistent, and chronic pediatric ITP has not been studied in detail but is assumed to be rare (2.4%).17,18 In adults, ∼18% to 38% of patients diagnosed with ITP have an underlying disease, comorbid condition, and/or comedication use, making the diagnosis of secondary ITP more probable.18,22-24 Cause and frequency of secondary ITP depend on demographic and socioeconomic factors. Infection-associated secondary ITP (eg, hepatitis C virus, Helicobacter pylori) especially exhibits strong geographic disparities.24-26 Known causes of secondary ITP include other systemic autoimmune disorders, primary or secondary immunodeficiency, infectious diseases, paraneoplastic syndrome (eg, lymphomas), and drug-dependent antibodies.3,16,24 Usually, assessment of the immunologic or nonimmunologic background of thrombocytopenia is difficult, and these mechanisms may induce thrombocytopenia simultaneously; indeed, this scenario is true for many forms of secondary ITP after a viral infection such as hepatitis C virus and HIV.27
Research pertaining to secondary ITP, particularly in pediatrics, is limited.15,17,18,20 We aimed to describe the natural history and clinical presentation of a cohort of children with secondary ITP and non-IT using data prospectively compiled over 15 years in the Pediatric and Adult Registry on Chronic ITP (PARC-ITP). Based on these findings, we proposed a diagnostic algorithm for more rapid identification of children with secondary ITP and non-IT, which may serve as a basis for further discussion and prospective trials.
Patients and methods
Ethics
PARC-ITP is an international prospective multicenter registry established in 2004. It was designed to collect clinical and laboratory information from children and adults with newly diagnosed primary ITP. The registry is a project of the Intercontinental Cooperative ITP Study Group (ICIS; www.itpbasel.ch), and its success is based on the voluntary participation of ICIS investigators. PARC-ITP collects data of patients with ITP at 95 institutions of 35 countries as of August 16, 2019.
All eligible patients are registered at the time of diagnosis. Data are assessed at 6 and 12 months after diagnosis and then yearly thereafter; they include demographic, ethnic, diagnostic, and clinical information as well as aspects of the management protocols. All included patients gave their informed consent. The study was conducted in accordance with the Declaration of Helsinki. The registry was approved by local review boards and ethical committees. The protocol, data registration process, and all registry documents are available via secure Internet access (www.parc-itp.net). Details on data acquisition, advantages, and limitations of the registry have been published elsewhere.28,29
Data acquisition
For this research, we extracted data from children and adolescents from PARC-ITP, for whom investigators reported “no” within 24 months of follow-up in response to the question “is primary ITP still assumed?” Patients suspected of having secondary ITP but with an unknown or unclear underlying disease (n = 57) were not evaluated because the etiology should have been known for the diagnosis of secondary ITP. Why these patients were suspected of having secondary ITP by the investigator is not known; this could be because of an atypical clinical presentation, nonresponse to first-line therapy, or because the question was misinterpreted. Patients whose diagnosis was again altered to primary ITP later at follow-up (n = 71) were excluded from the analysis. In addition, patients aged <3 months and patients aged >16 years were excluded (Figure 1).

Definitions
If not mentioned otherwise, standardized definitions were adopted.1 Age and initial data refer to the age and information collected at the time of diagnosis of ITP (day 0). The identification or switch to secondary ITP or non-IT reflects a diagnostic decision based on a subsequent dataset that reports a revision of the initial diagnosis (primary ITP) to secondary ITP or non-IT (at 6, 12, or 24 months after initial data collection). Follow-up “late” is the next available follow-up point after the revised diagnosis of secondary ITP or non-IT.
Secondary ITP was defined as encompassing “all forms of assumed immune-mediated thrombocytopenia except primary ITP.”1 Non-IT encompasses patients with thrombocytopenia of probably nonimmunologic causes such as bone marrow failure, malignancy, and congenital thrombocytopenia. The registry did not list or offer references regarding the accurate diagnosis of underlying diseases.
Platelet counts were recorded as values at the dates of follow-up visits, whereas information about bleeding and treatment were recorded as clinical data between 2 follow-up visits. Bleeding was defined as any event of hemorrhage, independent of the intensity or frequency of bleeding. The location of bleeding was reported for all bleeding events during the last observation period rather than per event. Corticosteroid therapy was defined as the administration of any form of corticosteroids irrespective of the dosage or route of administration. Comorbid conditions were defined as one or more of the following conditions at the time of initial registration and specifically queried by the registry: infectious diseases, autoimmune diseases (eg, rheumatoid arthritis, psoriasis, systemic lupus erythematosus), cardiovascular diseases, pulmonary disease, gastrointestinal diseases, endocrinologic diseases (eg, thyroid disease), cancer, and splenomegaly.
Statistical analysis
Descriptive statistics are presented as counts and frequencies for categorical data and mean (standard deviation) and median (interquartile range [IQR]) for metric variables. Overall P values correspond to Student t test (for means), Mann-Whitney U test (for medians), and χ2 or Fisher’s exact test when the expected frequencies were <5 in some cells. A P value <.05 was considered statistically significant. All evaluations were performed by using the statistical software R (R Foundation for Statistical Computing).
Results
A total of 3974 children with an initial diagnosis of primary ITP were registered in the PARC-ITP database between 2004 and 2019. Revisions to the diagnosis were reported for 241 children within 24 months of follow-up. Ultimately, 113 patients had an unequivocal diagnosis of secondary ITP or non-IT and were further analyzed (Figure 1). Geographical differences in the rate of secondary ITP/non-IT were not meaningful. Percentages of revised diagnosis, including lower and upper 95% confidence intervals, were as follows: South America, 5.6% (3.9-7.7); Eastern Europe, 3.9% (2.3-6.1); Africa, 3.4% (1.7-6); North America, 3.1% (1.7-5); Western Europe, 2.5% (1.5-3.9); Western Asia, 1.8% (0.7-4); and Eastern Asia, 0.8% (0.4-1.4).
Patients were analyzed in different groups according to the underlying causes (infection, autoimmune diseases, bone marrow disorders, malignancy, immunodeficiency, and drug use) (Tables 1 and 2). Information about etiology is provided in Figure 2.

Causes of secondary ITP or non-IT in children misdiagnosed with primary ITP. Infection: HIV, n = 5; Chagas disease, n = 1; herpes zoster, n = 1; no information, n = 47. Autoimmunity: systemic lupus erythematosus, n = 16; Evans syndrome, n = 11; autoimmune lymphoproliferative syndrome, n = 2; autoimmune bicytopenia (thrombocytopenia and neutropenia), n = 5; mixed connective tissue disease, n = 2; diabetes mellitus type 1, n = 1; scleroderma, n = 1; no information, n = 4. Malignancy: myelodysplastic syndrome, n = 3; Hodgkin lymphoma, n = 1; myeloproliferative disease, n = 2; no information, n = 1. Immunodeficiency: DiGeorge syndrome, n = 1; primary chronic granulomatous disease, n = 1; not determined, n = 2.
Causes of secondary ITP or non-IT in children misdiagnosed with primary ITP. Infection: HIV, n = 5; Chagas disease, n = 1; herpes zoster, n = 1; no information, n = 47. Autoimmunity: systemic lupus erythematosus, n = 16; Evans syndrome, n = 11; autoimmune lymphoproliferative syndrome, n = 2; autoimmune bicytopenia (thrombocytopenia and neutropenia), n = 5; mixed connective tissue disease, n = 2; diabetes mellitus type 1, n = 1; scleroderma, n = 1; no information, n = 4. Malignancy: myelodysplastic syndrome, n = 3; Hodgkin lymphoma, n = 1; myeloproliferative disease, n = 2; no information, n = 1. Immunodeficiency: DiGeorge syndrome, n = 1; primary chronic granulomatous disease, n = 1; not determined, n = 2.
Patient characteristics at initial presentation (day 0)
The initial characteristics of the patients with ITP eventually diagnosed with secondary ITP or non-IT in subsequent months are shown in Table 1. The median age in girls (9.2 years) was found to be significantly higher than in boys (4.2 years; P = .034). Patients with autoimmune disease–associated secondary ITP were older (12.4 years) than those with infection-associated secondary ITP (3.2 years; P < .001).
Four patients had a family history positive for thrombocytopenia; 2 of these patients were diagnosed during follow-up with systemic lupus erythematosus. In addition, 1 patient had DiGeorge syndrome, and 1 had autoimmune bicytopenia (ITP and autoimmune neutropenia). Comedications were reported in 14 patients and included anti-inflammatory drugs (n = 6), antimicrobial agents (n = 6), and nonsteroidal anti-inflammatory drugs (n = 2).
Initial diagnostic evaluation included a bone marrow puncture in 47 patients (42%). The findings of these punctures were not recorded in the registry; however, inclusion criteria requested consistency with the diagnosis of primary ITP. The rate of bone marrow puncture in patients with unchanged primary ITP was identical, with 1660 (42%) of 3920 evaluable children. Initial investigations also comprised serologic tests for HIV, hepatitis C virus, antiphospholipid antibody, antinuclear antibody (ANA), and antiplatelet antibody in 61 (4 positive), 62 (0 positive), 22 (0 positive), 44 (13 positive), and 16 (7 positive) patients, respectively, and Helicobacter pylori diagnostic test in 18 patients (1 positive). Of the 13 patients with positive ANAs, 12 were diagnosed later with autoimmune disease–associated secondary ITP; 4 of these patients also had antiplatelet antibodies. One patient with ANA and one with antiplatelet antibody had infection-associated secondary ITP. Of interest is that ANA was initially tested for 60% of patients having a later diagnosis of autoimmune-associated ITP and was only tested for 27% of all other patients in this cohort. Two patients with antiplatelet antibodies were diagnosed with aplastic anemia.
Patients with non-IT such as malignancy and aplastic anemia had significantly higher platelet counts at diagnosis (37 × 109/L) than those either with infection-associated (12 × 109/L; P = .026) or systemic autoimmune disease–associated (13 × 109/L; P = .016) secondary ITP. Bleeding manifestations were reported in 93 patients (82%), including 87 with cutaneous bleeding and 62 with mucosal bleeding, as well as one 9-year-old girl with muscle bleeding, diagnosed at follow-up with cancer. Platelet-enhancing treatment was given initially to 73 patients (65%); of these, 45 received immunoglobulins, 15 received corticosteroids, 11 received a combination of both immunoglobulin and corticosteroids, and 2 received second-line treatment.
The initial characteristics of patients with ITP diagnosed with secondary ITP compared with those of primary ITP patients are shown in Table 3.
Diagnosis of secondary ITP and non-IT
The diagnosis of secondary ITP or non-IT was reported for two-thirds (n = 73) of the patients using the 6-month follow-up questionnaire, for 18% of patients using the 12-month follow-up questionnaire, and for 18% of patients using the 24-month follow-up questionnaire. The characteristics reported to accompany the new diagnosis of secondary ITP or non-IT are shown in Table 2. Patients with autoimmune diseases had a significantly higher rate of ITP persisting longer than 6 months (60%) compared with those with infectious diseases (15%; P < .001).
Almost two-thirds of patients reported taking no platelet-enhancing treatment during the time period (6-12 months) preceding the revised diagnosis of secondary ITP or non-IT (n = 74). In the remaining 39 patients, treatment included corticosteroids (n = 21), intravenous immunoglobulin (IVIG) (n = 5), or both IVIG and corticosteroids (n = 8); 18 patients (46% of treated patients) received second-line treatments. The sustained need for platelet-enhancing drugs was higher in patients with an underlying autoimmune disease (62%) than in those with an infectious disease (19%; P < .001. A majority of patients did not exhibit bleeding in the same time period (n = 77). Types of bleeding seen in the remaining 36 patients included cutaneous bleeding (n = 26), mucosal bleeding (n = 36), and intracranial bleeding in 1 patient with aplastic anemia and 1 with HIV-associated ITP.
Follow-up “late”data
Follow-up “late” data were extracted from the first questionnaire after the change in diagnosis an additional 6 to 12 months later. Information was available for 76 (67%) of 113 children. At this point, 5 children had platelet values <20 × 109/L, 14 had platelet values between 20 and 100 × 109/L, and 44 patients had platelet values >100 × 109/L. The group of autoimmune-associated ITP had significantly more patients with chronic ITP (48%) than the group with infection-associated ITP (15%; P = .026). Sixty-two patients did not exhibit any bleeding, whereas patients with bleeding had cutaneous bleeding (n = 9) and/or mucosal bleeding (n = 12). Fifty-nine patients had received no treatment; the other patients had received IVIG (n = 2), corticosteroids (n = 10), or both (n = 2) and 4 patients had been administered second-line medications.
Infections
At this point, 36 patients were analyzed in this subgroup. The median age was 4.9 years (IQR: 2.9; 7.5 years). For the majority, the platelet count was in the normal range; 1 patient had a platelet count <20 × 109/L, and 2 patients had platelet counts between 20 and 100 × 109/L. For 10 patients with follow-up visits, no platelet count values were available. No bleeding was reported for 33 patients, and 2 patients experienced cutaneous bleeding. A watch-and-wait strategy was designated for 34 patients, 1 patient received corticosteroids, and 1 patient received second-line treatment.
Autoimmunity
In this subgroup, 26 patients were analyzed; the median age was 14.3 years (IQR: 8.3; 15.5 years), and the median platelet count was 109 × 109/L (IQR: 64; 217 × 109/L). Twelve patients had platelet counts >100 × 109/L, seven patients had platelet counts between 20 and 100 × 109/L (median: 68 × 109/L), and three patients had platelet counts <20 × 109/L. For 3 patients, no platelet count values were available. Seven patients (27%) showed bleeding, including 5 with cutaneous bleeding and 6 with mucosal bleeding. Platelet-enhancing drugs, including IVIG (n = 2), corticosteroids (n = 10), and the combination of both drugs (n = 1), were administered to 13 patients; 1 patient received anti-D immunoglobulin.
Malignancy (non-IT)
One of seven patients was lost to follow-up. The median age was 3.7 years (IQR: 2.6; 8.9 years), and the median platelet count was 193 × 109/L (IQR: 148; 255 × 109/L). Five patients had platelet counts >100 × 109/L and 1 patient had platelet counts between 20 and 100 × 109/L, the latter with mucosal bleeding.
Aplastic anemia (non-IT)
Follow-up information was available for 3 of 6 patients. The median age was 12.2 years (IQR: 10.3; 14.1 years), and the median platelet count was 76 × 109/L (IQR: 64; 131 × 109/L). Two patients had platelet counts between 20 and 100 × 109/L, and one patient had a platelet count >100 × 109/L. Two patients experienced bleeding, including 1 with cutaneous bleeding and 1 with both cutaneous and mucosal bleeding. One patient received IVIG.
Immunodeficiency
Information was available for 3 of 4 patients. The median age was 14.6 years (IQR: 8.6; 14.7 years), and the median platelet count was 107 × 109/L (IQR: 54; 155 × 109/L). One patient had a platelet count <20 × 109/L (1 × 109/L), and the other 2 patients had platelet counts >100 × 109/L. One patient experienced both cutaneous and mucosal bleeding, and no patients received any specific treatments.
Discussion
This study sought to assess children with a diagnosis of primary ITP that was revised to secondary ITP or to non-IT within 24 months of follow-up. A revision of the diagnosis of primary ITP was rare in our cohort (2.8%), and the main causes were almost equally infectious and autoimmune disorders. We assume that infectious diseases, especially acute infections such as Epstein-Barr virus, cytomegalovirus, and varicella-zoster virus, are underestimated in this analysis because this is one of the exclusion criteria for registration in PARC-ITP. In a chart review over 3 years at a clinic in Ankara, Turkey, Yenicesu et al30 found 10 (13%) of 75 ITP patients had positive serologic findings for Epstein-Barr virus, cytomegalovirus, or rubella. We did not identify any patient with inherited thrombocytopenia in our cohort, although such cases may mimic primary ITP and thus may be underrepresented.12,15,20,31-33 Because genetic screening for inherited etiologies of thrombocytopenia is now more readily available, rates of detection and diagnosis of these nonimmune platelet disorders will surely rise.5,20,31
The clinical characteristics of children with secondary infection–associated ITP seem to be very similar to those with primary ITP, possibly reflecting a similar disturbance in the immune system. Indeed, one hypothesis suggests that primary ITP is due to the cross-reactivity of antiplatelet antibodies and viral antigens (ie, antigenic mimicry).34 In a majority of children with primary ITP, infection occurred some weeks before the clinical manifestation of ITP.34-37 In contrast, the clinical characteristics of all other patients with secondary ITP and non-IT differ from those of children with primary ITP. In accordance with the literature, differences include older age, a predominance of female sex, greater frequencies of comorbidities, and positive family history for ITP, and reduced rates of bleeding at the initial diagnosis of ITP.15,16,18,19,21 Contrary to the expectation, initial platelet counts and the number of patients with profound thrombocytopenia (<20 × 109/L) were not significantly different between secondary and primary ITP19,21 ; however, patients with non-IT such as malignancy and aplastic anemia had significantly higher platelet counts at diagnosis than those either with infection-associated or autoimmune disease–associated secondary ITP. We assume that this finding reflects the more protracted disturbance of hematopoiesis compared with the rapid platelet destruction induced by antiplatelet antibodies. The bleeding frequency at initial diagnosis seems to be less often reported in patients with autoimmune disease than in those with an infectious disease or primary ITP; however, this finding was not statistically significant. No specific bleeding pattern was found to be distinctly indicative of secondary ITP in our cohort relative to primary ITP. The sustained need for platelet-enhancing drugs was significantly higher in patients with an underlying autoimmune disease than in patients with an infectious disease, and 60% and 20% of them were treated with second-line therapy, respectively. This outcome reflects previous findings in the literature, highlighting more first-line treatment failures in adult patients with secondary ITP.15,17,19,20
Investigators undertook a revision of the diagnosis of primary ITP in 65% of patients within the first 6 months, whereas a delayed diagnosis (>6-24 months) was reported in 50% of patients with aplastic anemia and 53% of patients with autoimmune diseases but only 22% of patients with infectious diseases and 14% of patients with malignancies. This variation may be caused by known insidious and delayed appearance of new symptoms and/or laboratory abnormalities in patients with aplastic anemia and autoimmune diseases. An algorithm might help shorten the delay or even improve the diagnosis initially.
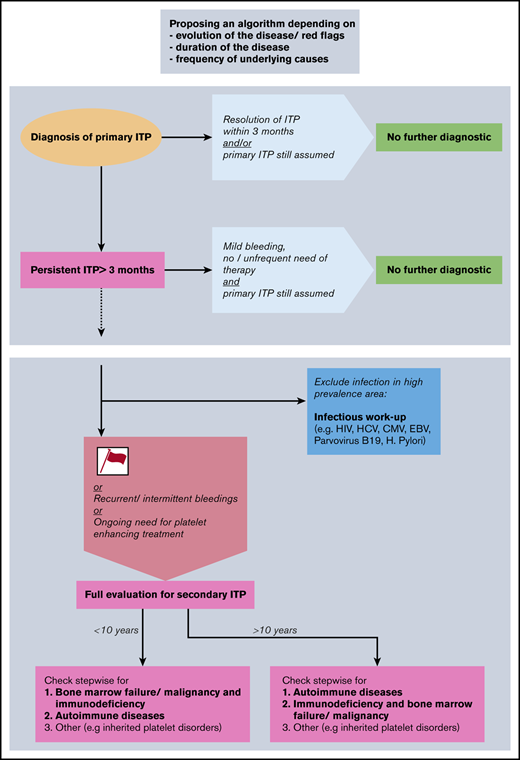
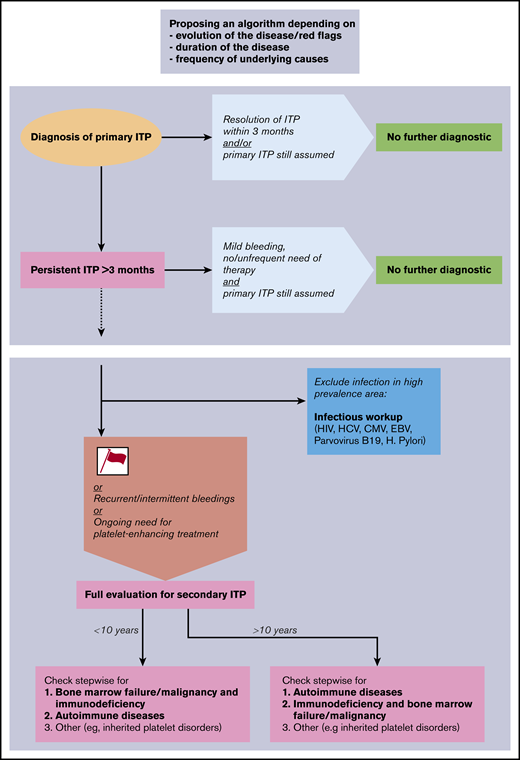
Given the results of our study, we propose a diagnostic algorithm for the evaluation of children with ITP (Figure 3). The proposed algorithm represents a clinical tool and not a laboratory or radiologic instruction guide. It is structured to be followed stepwise over time, includes red flags or risk factors, and prioritizes the sequences of diagnostic workup. The proposed algorithm may serve as a basis for further consensus discussions; the purpose and structure of other algorithms are presented in Table 4. Following our results and in accordance with existing literature,16,20,31 we propose the following clinical features to be early risk factors for secondary ITP or non-IT: female sex, age >10 years, moderate thrombocytopenia >20 but <100 × 109/L, positive family history for ITP, comorbidities, and nonresponse to first-line treatment. Because primary ITP is typically a self-limited condition in children, these risk factors are of greater importance if the disease persists for >3 to 6 months. We recommend conducting an expanded evaluation of patients with life-threatening and persistent bleeding requiring therapy, despite the fact that patients with the diagnosis of secondary ITP or non-IT do not seem to be at greater risk for bleeding. All patients with new symptoms or laboratory abnormalities during the disease course should be evaluated and do not appear in the algorithm, as the diagnosis is guided by the new findings.

Proposal of a diagnostic algorithm for pediatric secondary ITP and non-IT. Red flags (or risk factors) are defined as female sex, age >10 years, moderate thrombocytopenia >20 × 109/L, positive family history, comorbidities, and nonresponse to first-line treatment. CMV, cytomegalovirus; EBV, Epstein-Barr virus; HCV, hepatitis C virus.
Proposal of a diagnostic algorithm for pediatric secondary ITP and non-IT. Red flags (or risk factors) are defined as female sex, age >10 years, moderate thrombocytopenia >20 × 109/L, positive family history, comorbidities, and nonresponse to first-line treatment. CMV, cytomegalovirus; EBV, Epstein-Barr virus; HCV, hepatitis C virus.
Algorithms exist in the literature mostly with the aim to describe which analyses should be undertaken in case of chronic ITP but do not appear in a subsequent and informative order. This is the case for both the international ITP guidelines5 and the Italian guidelines.38 Recently, Consolini et al16 suggested a progressive algorithm with further investigations to be performed in patients with persistent and chronic ITP. The algorithm is driven by the specific examinations that should be undertaken and not by the possible differential diagnosis.
Limitations of our study include the use of narrow questionnaire-based information. For example, the PARC-ITP does not gather information on why secondary ITP was suspected and the steps of the diagnostic workup. The PARC-ITP reflects the actual diagnostic and clinical practices of a multinational group of volunteer investigators located worldwide. The registry does not recommend a diagnostic approach, and investigators are free to follow local or international guidelines. The questionnaire also does not query for details in cases of secondary ITP or non-IT, such as information regarding the infectious agent, specific therapy, or resolution of underlying disease. Epidemiologic statements must be carefully interpreted because the group of patients with secondary ITP and non-IT is small, and the data of patients registered in PARC-ITP reflect case-based and not population-based data, as described elsewhere.28,29
In summary, we described the rate of occurrence of a revised diagnosis of primary ITP into secondary ITP or non-IT in children recorded in a large, prospective, multicenter, international registry over 15 years. Infectious and systemic autoimmune disorders are the most frequent causes of secondary ITP, whereas oncologic disorders and bone marrow failure as differential diagnoses of ITP are extremely rare. This study may serve as a basis for designing prospective trials and for consensus discussions to establish diagnostic procedures in children with ITP.
Presented in abstract form at the 60th annual meeting of the American Society of Hematology, Orlando, FL, 29 November 2018.
Requests for data sharing may be submitted to the corresponding author (Alexandra Schifferli; e-mail: alexandra.schifferli@ukbb.ch).
Acknowledgments
The authors thank all patients and their families who offered their data for use in the PARC-ITP and the staff of the participating institutions. They highly appreciate the commitment of the ICIS investigators, who offered support by providing their patient data. The ICIS investigators are listed under www.itpbasel.ch/participants/. The authors are indebted to Verena Stahel, Caroline Asal Martin, and Monika Imbach for data administration and secretarial work, and they thank Andy Schötzau, Departement Biomedizin, University of Basel, for the statistical analysis. The authors thank Enago (www.enago.com) for the English language review.
The PARC-ITP is supported in part by grants from the Eduard Waeffler-Ludwig-Stiftung, Stiftung zur Förderung medizinischer und biologischer Forschung, and University Children’s Hospital Basel, University of Basel.
Authorship
Contribution: T.K., P.I., and A.S. designed and conducted the registry; and A.S., A.H., and T.K. analyzed and interpreted data, and wrote the paper; S.H., M.G.S., D.N., and M.M. reviewed the manuscript and provided content and editorial input; and all authors approved the final manuscript.
Conflict-of-interest disclosure: A.S. reports advisory board membership for Novartis and research funds from Amgen. M.M. serves as a consultant for Novartis, Amgen, and Rigel. T.K. reports research funds from Novartis and Amgen; and advisory board membership for UCB Biosciences. S.H. reports advisory board membership for Novartis. M.G.S. reports advisory board membership for Jazz Pharmaceuticals; honoraria from Novartis; and travel grants from Shire and Amgen. The remaining authors declare no competing financial interests.
Correspondence: Alexandra Schifferli, Department of Oncology/Hematology, University Children’s Hospital Basel, Spitalstr 33, 4031 Basel, Switzerland; e-mail: alexandra.schifferli@ukbb.ch.