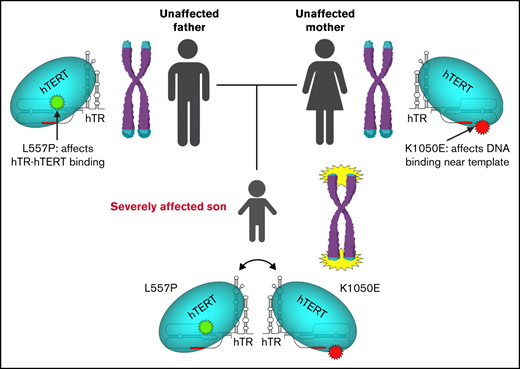
Key Points
A patient with a severe form of telomere biology disorder has compound heterozygous variants in TERT that interact functionally.
The functional interaction between alleles explains the much more extreme phenotype in the proband compared with his heterozygous parents.

Abstract
Telomere biology disorders (TBDs) are a spectrum of multisystem inherited disorders characterized by bone marrow failure, resulting from mutations in the genes encoding telomerase or other proteins involved in maintaining telomere length and integrity. Pathogenicity of variants in these genes can be hard to evaluate, because TBD mutations show highly variable penetrance and genetic anticipation related to inheritance of shorter telomeres with each generation. Thus, detailed functional analysis of newly identified variants is often essential. Herein, we describe a patient with compound heterozygous variants in the TERT gene, which encodes the catalytic subunit of telomerase, hTERT. This patient had the extremely severe Hoyeraal-Hreidarsson form of TBD, although his heterozygous parents were clinically unaffected. Molecular dynamic modeling and detailed biochemical analyses demonstrate that one allele (L557P) affects association of hTERT with its cognate RNA component hTR, whereas the other (K1050E) affects the binding of telomerase to its DNA substrate and enzyme processivity. Unexpectedly, the data demonstrate a functional interaction between the proteins encoded by the two alleles, with wild-type hTERT rescuing the effect of K1050E on processivity, whereas L557P hTERT does not. These data contribute to the mechanistic understanding of telomerase, indicating that RNA binding in one hTERT molecule affects the processivity of telomere addition by the other molecule. This work emphasizes the importance of functional characterization of TERT variants to reach a definitive molecular diagnosis for patients with TBD, and, in particular, it illustrates the importance of analyzing the effects of compound heterozygous variants in combination, to reveal interallelic effects.
Introduction
Telomeres are repeated DNA sequences capping the ends of chromosomes. In human stem cells, including those in the bone marrow, the enzyme telomerase maintains the length and integrity of telomeres.1,2 In patients with inherited mutations in the genes encoding telomerase or other proteins involved in telomere maintenance, telomeres become abnormally short, leading to a spectrum of disorders known collectively as telomere biology disorders (TBDs),3-5 which often feature bone marrow failure (BMF). Short or dysfunctional telomeres lead to activation of a DNA damage response and cellular senescence or apoptosis, which reduces the replicative capacity of hematopoietic stem cells, leading to progressive BMF.6
The first of these diseases linked to short telomeres was dyskeratosis congenita (DC); the gene known to be mutated in a subset of patients with DC, DKC1,7 was shown to encode a protein (dyskerin) that stabilizes the RNA component of telomerase, hTR,8 and is an integral component of active telomerase.8,9 Subsequently, mutations in the genes TERC10 (encoding hTR) and TERT11 (encoding hTERT, the catalytic protein subunit of telomerase), were also found to be mutated in patients with DC; there are now at least 15 other genes implicated in TBD inheritance, all of which affect telomere integrity when mutated.4,12 -15 TBDs are variable and multisystem disorders, with clinical manifestations that include pulmonary fibrosis, liver cirrhosis, gastrointestinal symptoms, dental abnormalities, and predisposition to malignancies.3,4 In addition to those features, severe forms of the disease include Hoyeraal-Hreidarsson syndrome, characterized by growth restriction, microcephaly, cerebellar hypoplasia, immunodeficiency and developmental delay,16,17 and Revesz syndrome, which is characterized by cerebral calcifications and exudative retinopathy.18,19
The catalytic subunit of human telomerase, hTERT, is a reverse transcriptase that interacts stably with its template-containing RNA subunit, hTR.20,21 hTERT catalyzes the addition of nucleotides to the 3′ end of a single-strand telomeric DNA substrate; when the end of the short template sequence is reached, the RNA molecule translocates relative to the DNA molecule, positioning the substrate for another round of addition.22 Telomerase is therefore capable of adding many copies of the telomere repeat sequence 5′-TTAGGG-3′ to a single DNA molecule, a process known as repeat addition processivity.23 The molecular basis of the unique mechanism underlying telomerase activity and processivity is beginning to be understood,24,25 but many details remain to be elucidated.
Disease-associated mutations in TERT are found throughout the protein-coding region of the gene, including in the protein domains responsible for reverse transcriptase activity, RNA binding, DNA binding, and localization of telomerase to telomeres.26 Most disease-causing TERT mutations are inherited in an autosomal dominant manner, but a small number of severely affected patients have biallelic mutations, resulting in autosomal recessive disease (supplemental Table 1).5 TERT mutations can display highly variable penetrance within families, with respect to both the degree of telomere shortening and the clinical phenotype.27-29 TBDs also demonstrate marked anticipation (ie, an increase in severity of the phenotype and earlier onset of disease in subsequent generations), due to the inheritance of shorter telomeres with each generation.30,31 Furthermore, in silico algorithms show very limited ability to discriminate between benign or deleterious variants in TERT.32 These factors often make it challenging to assign pathogenicity to newly identified variants in TERT and other TBD genes, necessitating functional analysis to reach a definitive molecular diagnosis.
Although many TERT variants have been demonstrated to impact telomerase activity, detailed molecular analyses of the impact of disease-associated mutations on aspects of telomerase biochemistry are more limited.33-37 Such analyses can provide a rich source of information for understanding telomerase biochemical mechanisms, which will in turn enlighten future attempts to assign pathogenicity to novel variants. We have constructed a refined structural model for the catalytic core of human telomerase based on a recently reported cryo–electron microscopy (cryo-EM) structural model38 and used it to conduct molecular modeling, enabling prediction of the impacts on telomerase function imparted by disease-associated TERT variants. We identified a family in which the proband was severely affected by Hoyeraal-Hreidarsson syndrome from infancy, and carried compound heterozygous variants in TERT (the previously described K1050E39 and novel variant L557P). Molecular dynamic modeling predicts that L557P impacts binding of hTERT to its RNA subunit, and that K1050E results in a change in hTR-DNA duplex stability, affecting DNA binding; we validated both predictions with biochemical analyses. Thus, the proband in this family carries 2 variants that affect different biochemical properties of telomerase.
Compound heterozygous disease mutations are rare in TERT-associated TBDs, but provide a unique opportunity to interrogate complex interactions imparted by different alleles on each other. Most reported disease-associated TERT variants are heterozygous, and although it is well established that a completely inactive version of hTERT can act in a dominant negative fashion to impair activity of the wild-type (WT) enzyme,40-42 this has not been described so far for any hypomorphic disease-associated TERT variant.27,30,43,44 Similarly, the only compound heterozygous TERT variants to have been functionally characterized together demonstrated activity that was intermediate between the two variants individually,45 indicating a lack of functional interaction. In contrast, by quantitatively analyzing telomerase processivity, we demonstrate herein a functional interaction between the two mutated hTERT proteins, resulting in a much more severe phenotype in the proband than either of his asymptomatic heterozygous parents.
Materials and methods
Subjects and samples
The proband in this study presented at 8 months of age to Queensland Children’s Hospital (Brisbane, QLD, Australia). Exome sequencing of a 57-gene inherited bone marrow panel was performed on peripheral blood DNA from the proband,46 his parents, and paternal grandmother. This research was approved by the Peter Mac Ethics Committee (HREC/13/MH/326 and HREC/17/PMCC/163), and all participants provided written informed consent.
Telomere length analysis
Telomere flow-fluorescence in situ hybridization (Flow-FISH) was performed with a published protocol47 using mononuclear cells isolated from peripheral blood. Calculation of relative telomere length of the patient’s mononuclear cells was performed by comparing the fluorescence of these cells with the tetraploid CCRF-CEM cell line,47 and the results were expressed as a percentage. Values obtained from 240 healthy individuals show the normal percentiles for different age groups.
Telomere length analysis
A previously described monochrome multiplex quantitative polymerase chain reaction (qPCR) telomere length assay48,49 was used, incorporating quantitative PCR with telc and telg telomere PCR primers (supplemental Table 2) and albu and albd single-copy gene PCR primers. Values obtained from 240 healthy individuals show the normal percentiles for different age groups.
Telomere length by Southern blot analysis
Telomere length was analyzed by electrophoresis of telomeric restriction fragments on an agarose gel and hybridization with a radiolabeled telomeric probe, as described.50
Structural model generation
Because of loop flexibility, the hTERT cryo-EM structure (Protein Data Bank [PBD]: 7BG9) is missing experimental density for amino acids 101 to 124, 180 to 320, and 416 to 443.38 Thus, a homology model of hTERT was generated by using Modeler v10.051,52 and the hTERT cryo-EM structure of hTERT as the model template.38 The nucleic acids from the cryo-EM structure were merged into this model. hTR RNA residues 238 to 248 and 322 to 333 were excluded because they did not interact with the hTERT protein. To be consistent with our experimental methods, the telomeric DNA sequence TTAGGG had to be extended to 3 repeats. The Tetrahymena thermophila TERT cryo-EM structure (PDB: 6D6V)53 includes an experimentally defined 3-repeat DNA substrate and was therefore used to position the DNA in our model. To create the mutant models (L557P and K1050E), the residues were selected, mutated using PyMOL,54 and minimized as described next.
Molecular dynamics simulation
All molecular dynamics simulations were performed in the program GROMACS55 (release 2021.2), using the AMBER99SB-ILDN force field56 and the TIP3P water model.57,58 The telomerase model was positioned in a dodecahedral water box with 10-Å padding on all sides. Sodium and chloride ions were added to neutralize the charge of the system, and then additional sodium and chloride ions were added to reach physiological conditions (150 mM). Before running the production runs, the system was energy minimized by using the steepest descent algorithm, until the maximum force in the system was less than 500 kJ/mol per nanometer. A subsequent constant number of particles, volume, and temperature equilibration was performed for 100 ps at 310 K. Two 10 ns (constant number of particles, pressure, and temperature) production runs were conducted at 310 K for the WT, hTERTL557P, and hTERTK1050E structures.
The root mean square deviation and root mean square fluctuation of the residues in WT, hTERTL557P, and hTERTK1050E were calculated with GROMACS and compared. To identify representative conformations within the simulations, all trajectories in each simulation were clustered by using the GROMOS algorithm.59 Hydrogen bonding and ionic bonding in the trajectory were quantified by using the program VMD,60 with the geometric definition of a hydrogen bond, <3.5 Å between hydrogen bond donor and acceptor and an angular cutoff of 30°, used for quantification. Statistical analyses were performed in GraphPad Prism and figures were created using PyMOL.54
Overexpression and purification of human telomerase
The human TERT gene,42 driven by a cytomegalovirus promoter was cloned into plasmid pcDNA3.1(+) (Invitrogen) with an N-terminal 6×FLAG tag. The TERC gene under a U3 promoter and DKC1 under a cytomegalovirus promoter were cloned into a single plasmid in vector pcDNA3.1(+). TERT variants 1670T>C (encoding L557P) and 3148A>G (encoding K1050E) were introduced into the FLAG-hTERT construct using the Takara In-Fusion HD Cloning Plus system according to the manufacturer’s recommendations, with the DNA changes incorporated into the TERT PCR primers (primer sequences listed in supplemental Table 2). The template plasmid was removed by digestion with 100 U of DpnI (New England Biolabs). All constructs were sequenced over the entire TERT gene to ensure that only the variant of interest was introduced. The telomerase complex was expressed in HEK293T cells and immunopurified as described previously.61
Western blot analysis
To determine the amount of hTERT recovered during immunopurification (IP), lysates and IP eluates were subjected to Western blot analysis and probed with a mouse monoclonal primary antibody (anti-FLAG; F3165; Sigma-Aldrich), as described.62
Direct telomerase activity assay
Relative telomerase activity was quantitated with a direct (ie, non-PCR) assay, as described.61 In brief, equal amounts of immunopurified telomerase (based on hTERT quantitation by western blot analysis) were used to extend a telomeric oligonucleotide primer in the presence of α-32P-dGTP and analyzed on acrylamide sequencing gels.
Northern dot blot analysis of telomerase RNA
The ability of hTERT to bind to hTR was determined by using northern dot-blot analysis to quantitate hTR recovery after IP against hTERT, as described.61
Telomerase-DNA binding assay
The equilibrium binding constant (KD) between telomerase and its telomeric DNA substrate was determined with a pulldown assay with a biotinylated DNA substrate used as described.61
Further details of published methods (Flow-FISH telomere length, qPCR telomere length, Southern blot telomere length, telomerase purification, and telomerase functional assays) are available in the supplemental Material.
Results
Clinical presentation and genetic analysis of proband and family members
An 8-month-old boy born to nonconsanguineous parents (Figure 1A) presented with petechiae, microcephaly, leucoplakia, brain stem and cerebellar hypoplasia, retinopathy, and subtle dysmorphic features (hypertelorism, low-set ears, and prominent nasal bridge). He had a history of intrauterine growth restriction and had required repair of bilateral inguinal hernias and a right orchidopexy at age 8 weeks. At 8 months, his bone marrow was moderately hypocellular with dysplastic features in erythroid and megakaryocytic lineages; there was no evidence of clonal progression on repeat examination 6 months later, and the World Health Organization Criteria for Myelodysplastic Syndrome63 were not met, particularly in the presence of an inherited BMF syndrome. At 15 months of age he underwent an allogeneic hematopoietic stem cell transplant (HSCT) for transfusion-dependent BMF. The initial matched unrelated donor (MUD) HSCT resulted in secondary graft failure; a subsequent MUD HSCT 3 months later was successful, but resulted in significant posttransplant gastrointestinal toxicity and stricturing, despite minimally intensive conditioning regimens for both transplants. At this writing, the proband is aged 4, has gross developmental delay, is fed parenterally, and has recently developed hepatopulmonary syndrome. The clinical presentation of the proband, together with telomere length and genetic and functional analysis described in the next sections, led to a diagnosis of Hoyeraal-Hreidarsson syndrome, with features of Revesz syndrome.
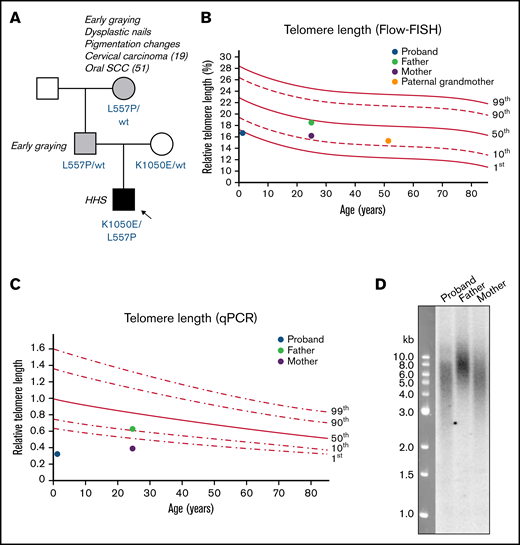
Family pedigree and telomere length measurements of patient with Hoyeraal-Hreidarsson syndrome. (A) Pedigree of family in which the proband has compound heterozygous variants in TERT. Amino acid changes resulting from the TERT variants are labeled in blue. Arrow and black shaded box indicate the proband with Hoyeraal-Hreidarsson syndrome; gray shading indicates those with mild DC features. (B) Relative telomere length (as a percentage of that of the control 4n cell line CCRF-CEM) according to age, measured by Flow-FISH. Lines represent the 1st, 10th, 50th, 90th, and 99th percentiles of telomere length of 240 healthy controls. (C) Relative telomere length according to age, measured by qPCR. Lines represent the 1st, 10th, 50th, 90th, and 99th percentiles of telomere length of 240 healthy controls. (D) Telomere length in the proband and his parents, measured by Southern blot analysis with a probe against telomeric DNA.
Family pedigree and telomere length measurements of patient with Hoyeraal-Hreidarsson syndrome. (A) Pedigree of family in which the proband has compound heterozygous variants in TERT. Amino acid changes resulting from the TERT variants are labeled in blue. Arrow and black shaded box indicate the proband with Hoyeraal-Hreidarsson syndrome; gray shading indicates those with mild DC features. (B) Relative telomere length (as a percentage of that of the control 4n cell line CCRF-CEM) according to age, measured by Flow-FISH. Lines represent the 1st, 10th, 50th, 90th, and 99th percentiles of telomere length of 240 healthy controls. (C) Relative telomere length according to age, measured by qPCR. Lines represent the 1st, 10th, 50th, 90th, and 99th percentiles of telomere length of 240 healthy controls. (D) Telomere length in the proband and his parents, measured by Southern blot analysis with a probe against telomeric DNA.
Germline mutational analysis of a 57-gene inherited BMF panel identified compound heterozygous missense variants in the TERT gene (NM_198253.2): a maternally inherited c.3148A>G;p(Lys1050Glu) allele, and a paternally inherited c.1670T>C;p(Leu557Pro) allele.46 Neither variant is present in The Genome Aggregation Database. Both parents were heterozygous carriers of their respective alleles and were healthy, with no hematological or pulmonary defects detectable. The proband’s father and paternal grandmother (who also carried a heterozygous c.1670T>C;p[Leu557Pro] allele) both experienced premature hair graying, and the paternal grandmother also displayed some skin pigmentation changes and dysplastic toenails; she had been diagnosed with cervical carcinoma at age 19 and oral squamous cell carcinoma at age 51, though she was a smoker.
Telomere length analyses of proband and family members
At age 8 months, the telomere length of peripheral blood mononuclear cells from the proband was low (∼1st percentile), as measured by a clinically accredited Flow-FISH assay (Figure 1B). His father had normal-length telomeres, and his mother and paternal grandmother both had telomeres on the low end of normal (just above the 10th percentile). Because the Flow-FISH assay has limitations related to the fragility of unfixed fresh blood cells,64 the relative telomere lengths of the proband and his parents were confirmed with 2 techniques that instead use purified genomic DNA. Both qPCR (Figure 1C) and Southern blot analysis (Figure 1D) confirmed that the proband’s average telomere length was shorter than that of both parents, despite his young age, and he had a greater proportion of very short telomeric fragments detectable on the Southern blot.
Modeling the structural impacts of each hTERT variant predicts different biochemical defects
The hTERT protein comprises 4 distinct functional domains (Figure 2A). The paternally inherited L557P variant is located in the RNA-binding domain, within the very highly conserved T-motif, which is known to form an extended pocket that interacts with the telomerase RNA subunit in several species.65-68 The K1050E variant is located in the C-terminal extension of hTERT, which we and others have demonstrated plays a major role in binding to the DNA substrate of telomerase.36,37,69 To model the structural impact of these variants on hTERT, we constructed a structural model of hTERT, using the model derived from the recent cryo-EM data as a starting point,38 and then modified our model to include a 3-repeat fully telomeric DNA substrate (Figure 2B).
![Molecular modeling predicts that L557P affects hTERT binding to hTR, whereas K1050E affects DNA binding. (A) Schematic of the secondary structure of hTERT, showing the 4 major functional domains: telomerase essential N-terminal (TEN) domain, RNA binding domain (RBD), reverse transcriptase (RT) domain, and C-terminal extension (CTE) domain. The alignments82 show the degree of conservation of the regions surrounding L557P and K1050E in 9 species, including vertebrates, a plant, ciliated protozoa, and yeast: hTERT, human; mTERT, Mus musculus; aTERT, Arabidopsis thaliana; OtTERT, Oxytricha trifallax; Eap123, Euplotes aediculatus; TtTERT, Tetrahymena thermophila; ScEst2p, Saccharomyces cerevisiae; CaTERT, Candida albicans; SpTRT1p, Schizosaccharomyces pombe. Identical positions are indicated in green; positions showing amino acid similarity are highlighted in gray. (B) Model of WT hTERT used for molecular dynamics simulations. Protein, RNA, and DNA are gray, magenta, and blue, respectively. Locations of investigated mutations, L557 and K1050, are represented as space-filling atoms and colored in green and red, respectively. (C) Effect of L557P (green spheres): close-up view of the shift in position of the 482-to-488 helix relative to hTR in hTERTL557P (right) compared with WT hTERT (left). The change in hydrogen bonding between the 482-to-488 loop and hTR is quantified in panel D; data are the mean ± standard error of the mean (SEM; n = 3). *P = .0147, by 2-tailed t test. (E) Effect of K1050E: in hTERTwt (gray, left), K1050 (red) forms a hydrogen bond with hTR (magenta) at adenine-55. This hydrogen bond is lost in the hTERTK1050E protein (right) (quantified in panel F; data are the mean ± SEM [n = 3]; **P = .0074, by 2-tailed t test). In the hTERTK1050E simulation, the DNA substrate (residues 11-18) is shifted away from the thumb loop (hTERT residues 957-965; wheat) compared with WT and is accompanied by a reduction in hydrogen bonding between the thumb loop and DNA in hTERTK1050E compared with WT (quantified in panel G; data are the mean ± SEM [n = 3]; *P = .0461, by 2-tailed t test). Hydrogen bonds are yellow dashed lines (E), and the distance between a protein residue and the nearest nucleotides (outside hydrogen bonding distances) are indicated by double-headed black arrows (E; right).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/12/10.1182_bloodadvances.2022007029/1/m_advancesadv2022007029f2.png?Expires=1769375554&Signature=yF5E40R6HNUA8GAEXhmxCkSSFmvT59fz8sWN8fITKisx-DimfRQlX4K6rbMzFGTU-GaJ0CQjnZMCLz3TfDgulE8eP-R7qybJDzKw6dxe54dGzWf~NAMuI93VfbSnoReB~mhCx8elhyCTyJWx3kf3wOEvimDL62Xm2IcZGe8LTHRsZSlao8Xz~b7jn9~Yg6Fo8aEytBnpOlEvkImY5ksyVqAUy3bf-SsUFlj0daqRZy6y83G9k0-yZ9qUIqHxfRMUzkT4fy4X2OPrEsIYlh533YGI6CNEl1zwbVeR-tivoByuBW4irlGubS~TVScNWsxJ0t9JcQ4xw89b~Cpvv0AFAQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Molecular modeling predicts that L557P affects hTERT binding to hTR, whereas K1050E affects DNA binding. (A) Schematic of the secondary structure of hTERT, showing the 4 major functional domains: telomerase essential N-terminal (TEN) domain, RNA binding domain (RBD), reverse transcriptase (RT) domain, and C-terminal extension (CTE) domain. The alignments82 show the degree of conservation of the regions surrounding L557P and K1050E in 9 species, including vertebrates, a plant, ciliated protozoa, and yeast: hTERT, human; mTERT, Mus musculus; aTERT, Arabidopsis thaliana; OtTERT, Oxytricha trifallax; Eap123, Euplotes aediculatus; TtTERT, Tetrahymena thermophila; ScEst2p, Saccharomyces cerevisiae; CaTERT, Candida albicans; SpTRT1p, Schizosaccharomyces pombe. Identical positions are indicated in green; positions showing amino acid similarity are highlighted in gray. (B) Model of WT hTERT used for molecular dynamics simulations. Protein, RNA, and DNA are gray, magenta, and blue, respectively. Locations of investigated mutations, L557 and K1050, are represented as space-filling atoms and colored in green and red, respectively. (C) Effect of L557P (green spheres): close-up view of the shift in position of the 482-to-488 helix relative to hTR in hTERTL557P (right) compared with WT hTERT (left). The change in hydrogen bonding between the 482-to-488 loop and hTR is quantified in panel D; data are the mean ± standard error of the mean (SEM; n = 3). *P = .0147, by 2-tailed t test. (E) Effect of K1050E: in hTERTwt (gray, left), K1050 (red) forms a hydrogen bond with hTR (magenta) at adenine-55. This hydrogen bond is lost in the hTERTK1050E protein (right) (quantified in panel F; data are the mean ± SEM [n = 3]; **P = .0074, by 2-tailed t test). In the hTERTK1050E simulation, the DNA substrate (residues 11-18) is shifted away from the thumb loop (hTERT residues 957-965; wheat) compared with WT and is accompanied by a reduction in hydrogen bonding between the thumb loop and DNA in hTERTK1050E compared with WT (quantified in panel G; data are the mean ± SEM [n = 3]; *P = .0461, by 2-tailed t test). Hydrogen bonds are yellow dashed lines (E), and the distance between a protein residue and the nearest nucleotides (outside hydrogen bonding distances) are indicated by double-headed black arrows (E; right).
Molecular modeling predicts that L557P affects hTERT binding to hTR, whereas K1050E affects DNA binding. (A) Schematic of the secondary structure of hTERT, showing the 4 major functional domains: telomerase essential N-terminal (TEN) domain, RNA binding domain (RBD), reverse transcriptase (RT) domain, and C-terminal extension (CTE) domain. The alignments82 show the degree of conservation of the regions surrounding L557P and K1050E in 9 species, including vertebrates, a plant, ciliated protozoa, and yeast: hTERT, human; mTERT, Mus musculus; aTERT, Arabidopsis thaliana; OtTERT, Oxytricha trifallax; Eap123, Euplotes aediculatus; TtTERT, Tetrahymena thermophila; ScEst2p, Saccharomyces cerevisiae; CaTERT, Candida albicans; SpTRT1p, Schizosaccharomyces pombe. Identical positions are indicated in green; positions showing amino acid similarity are highlighted in gray. (B) Model of WT hTERT used for molecular dynamics simulations. Protein, RNA, and DNA are gray, magenta, and blue, respectively. Locations of investigated mutations, L557 and K1050, are represented as space-filling atoms and colored in green and red, respectively. (C) Effect of L557P (green spheres): close-up view of the shift in position of the 482-to-488 helix relative to hTR in hTERTL557P (right) compared with WT hTERT (left). The change in hydrogen bonding between the 482-to-488 loop and hTR is quantified in panel D; data are the mean ± standard error of the mean (SEM; n = 3). *P = .0147, by 2-tailed t test. (E) Effect of K1050E: in hTERTwt (gray, left), K1050 (red) forms a hydrogen bond with hTR (magenta) at adenine-55. This hydrogen bond is lost in the hTERTK1050E protein (right) (quantified in panel F; data are the mean ± SEM [n = 3]; **P = .0074, by 2-tailed t test). In the hTERTK1050E simulation, the DNA substrate (residues 11-18) is shifted away from the thumb loop (hTERT residues 957-965; wheat) compared with WT and is accompanied by a reduction in hydrogen bonding between the thumb loop and DNA in hTERTK1050E compared with WT (quantified in panel G; data are the mean ± SEM [n = 3]; *P = .0461, by 2-tailed t test). Hydrogen bonds are yellow dashed lines (E), and the distance between a protein residue and the nearest nucleotides (outside hydrogen bonding distances) are indicated by double-headed black arrows (E; right).
Molecular dynamics simulations of hTERT with the L557P variant suggested that there is a reduced interaction of the hTERT protein with hTR residues 178 to 182 and 105 to 108, in the pseudoknot region of hTR. L557 resides at the base of an α-helix that connects to the T-motif β-hairpin (Figure 2C). This α-helix is adjacent to the loop containing residues 306 to 321, which is, in turn, adjacent to an α-helix formed by residues 482 to 488. In the WT protein, this α-helix fits into the groove of the triple helix70 of the hTR pseudoknot (Figure 2C, left; RNA in magenta). The L557P substitution caused the 482 to 488 α-helix to rotate from its optimal orientation in this hTR groove, which led to a reduction in the number of hydrogen bonds between the protein and hTR (Figure 2C, right). Overall, the average number of hydrogen bonds decreased approximately threefold in the L557P simulation relative to WT (Figure 2D), and the time of occupancy (a measure of stability) of many of these bonds also decreased. Thus, we predicted that this mutant would result in reduced affinity of the hTERT protein to hTR.
In our structural model, K1050 resides within the C-terminal extension of hTERT, with its side chain in proximity to the DNA-RNA duplex in the template region of hTR (Figure 2E, left). The reversal in amino acid side-chain charge of hTERTK1050E completely disrupted the ability of this residue to hydrogen bond to hTR (Figure 2E, right; Figure 2F). This changed the conformation of the DNA-RNA duplex, with the DNA substrate shifted farther away from the thumb loop (residues 957-965) in the hTERTK1050E simulation relative to WT (Figure 2E; supplemental Figure 1). The shift seen in these simulations caused a reduction in the amount of hydrogen bonding between the thumb loop and DNA (Figure 2G). The thumb loop has been proposed to contribute to single-strand DNA retention during DNA-RNA duplex dissociation and template translocation,69 and a nearby mutation in the C terminus of hTERT also affected thumb loop interactions with DNA and reduced telomerase processivity.36 Thus, altering the interaction of DNA with the thumb loop is predicted to change DNA-RNA duplex dynamics and reduce enzyme repeat addition processivity.
L557P affects telomerase activity, whereas K1050E reduces processivity
L557P is a novel hTERT variant, whereas K1050E has been reported in 2 individuals with inherited pulmonary fibrosis.39,71 In one of those studies,39 telomerase activity of in vitro–reconstituted K1050E telomerase was not significantly different from WT, whereas a later study reported that K1050E reduces activity by ∼50%.37 To resolve this discrepancy and determine the effects of each variant individually and together on telomerase enzymatic properties, we transfected HEK293T cells with plasmids encoding each hTERT variant, together with a plasmid expressing both hTR and dyskerin to reconstitute the telomerase complex in a human cell culture system. Under these conditions, the exogenous telomerase complex exceeds that present endogenously in HEK293T cells by ≥50-fold,61 and the small amount of endogenous telomerase is removed by use of a FLAG immunoprecipitation of the FLAG-tagged exogenous enzyme. To recapitulate the compound heterozygous genotype of the proband, a 1:1 mixture of L557P and K1050E hTERT plasmids was transfected, and a 1:1 mixture of WT with each mutant hTERT recapitulated the heterozygous parental genotypes. Both the L557P and K1050E hTERT proteins were present at levels approximately equal to WT hTERT (Figure 3A), indicating no effect on protein stability by either mutation.
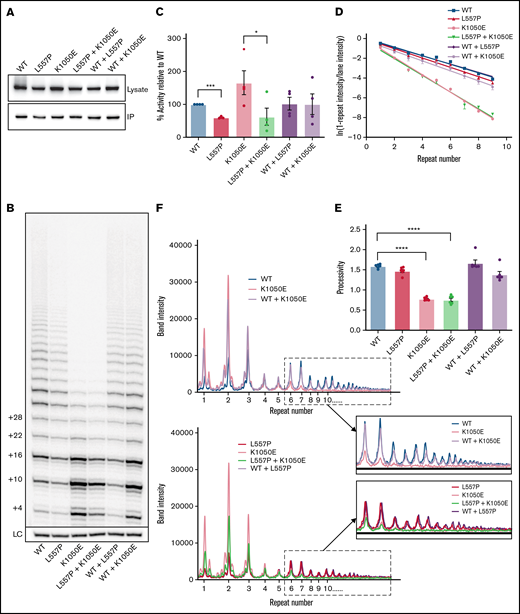
The effects of L557P and K1050E on telomerase activity. (A) Western blot analyses of HEK293T cell lysates expressing the indicated hTERT variants (top) and the corresponding samples purified by IP (bottom). Blots were probed with an anti-FLAG antibody to detect exogenous FLAG-tagged hTERT. (B) Direct activity assay showing extension of a telomeric DNA primer in vitro in the presence of radiolabeled 32P-dGTP, for WT telomerase and the indicated variants, expressed individually or together. The number of nucleotides added to the primer is shown on the left. LC: 32P-labeled 30-mer oligonucleotide included as a control for recovery and loading. (C) Quantitation of total telomerase activity relative to WT for the indicated variants or combinations. Data are the mean ± standard error of the mean (SEM; n = 4). *P = .042; ***P = .0003, by repeated-measures 1-way analysis of variance (ANOVA), followed by Dunnett’s post hoc test. (D) Calculation of the amount of telomerase processivity75 for the indicated hTERT variants and combinations. For each telomere repeat added by telomerase, the logs of the “fraction of products left behind” (ie, dissociated from the enzyme) were plotted against the repeat number, and the plot was fitted by linear regression, with the slope inversely proportional to processivity. Each data point represents the mean ± SEM (n = 6). (E) Processivity values (defined as −0.693/m, where m is the slope of the line) were calculated from the 6 individual experiments shown in panel D. Data are the mean ± SEM (n = 6). ****P < .0001, by repeated measures 1-way ANOVA, followed by Dunnett’s post hoc test. (F) Line graphs of the intensities of bands in the indicated lanes of the gel in panel B. WT and K1050E telomerase, expressed individually or together (top). L557P and K1050E telomerase, expressed individually or together (bottom). Magnification of the boxed regions of the plots, showing repeat 6 and higher (inset).
The effects of L557P and K1050E on telomerase activity. (A) Western blot analyses of HEK293T cell lysates expressing the indicated hTERT variants (top) and the corresponding samples purified by IP (bottom). Blots were probed with an anti-FLAG antibody to detect exogenous FLAG-tagged hTERT. (B) Direct activity assay showing extension of a telomeric DNA primer in vitro in the presence of radiolabeled 32P-dGTP, for WT telomerase and the indicated variants, expressed individually or together. The number of nucleotides added to the primer is shown on the left. LC: 32P-labeled 30-mer oligonucleotide included as a control for recovery and loading. (C) Quantitation of total telomerase activity relative to WT for the indicated variants or combinations. Data are the mean ± standard error of the mean (SEM; n = 4). *P = .042; ***P = .0003, by repeated-measures 1-way analysis of variance (ANOVA), followed by Dunnett’s post hoc test. (D) Calculation of the amount of telomerase processivity75 for the indicated hTERT variants and combinations. For each telomere repeat added by telomerase, the logs of the “fraction of products left behind” (ie, dissociated from the enzyme) were plotted against the repeat number, and the plot was fitted by linear regression, with the slope inversely proportional to processivity. Each data point represents the mean ± SEM (n = 6). (E) Processivity values (defined as −0.693/m, where m is the slope of the line) were calculated from the 6 individual experiments shown in panel D. Data are the mean ± SEM (n = 6). ****P < .0001, by repeated measures 1-way ANOVA, followed by Dunnett’s post hoc test. (F) Line graphs of the intensities of bands in the indicated lanes of the gel in panel B. WT and K1050E telomerase, expressed individually or together (top). L557P and K1050E telomerase, expressed individually or together (bottom). Magnification of the boxed regions of the plots, showing repeat 6 and higher (inset).
Immunopurified telomerase was analyzed with a direct primer-extension assay, to measure the ability of the enzyme to extend telomeric DNA. The L557P variant reduced activity by ∼50% (Figure 3B-C). K1050E resulted in a marked reduction in telomerase processivity, indicated by an increase in the relative intensity of shorter extension products (Figure 3B), as previously reported.37 The overall intensity of K1050E extension products was higher than those produced by WT telomerase (Figure 3C); however, this was most likely an effect of the reduction in processivity, because earlier dissociation of the enzyme from each extended DNA molecule would allow greater enzyme turnover in an in vitro assay.32,69,72,73 Variations in the amount of enzyme turnover depending on reaction conditions may be the reason that previous analyses have provided conflicting results regarding the effect of K1050E on total activity levels.37,39 This would not reflect the situation in vivo, where the effective DNA concentration is lower; indeed, it has been demonstrated that an hTERT mutation with a similar defect in processivity but increased apparent in vitro activity caused by higher enzyme turnover is defective in telomere maintenance in cells,69 showing that telomerase processivity most likely plays a more important role than enzyme turnover in telomere lengthening in cells.
Use of a direct assay (rather than the commonly used PCR-based TRAP assay39 ) allowed us to quantitate enzyme processivity precisely, showing that K1050E leads to an ∼50% reduction (P < .0001), whereas the processivity of L557P is not significantly different from that of the WT (P = .084; Figure 3D-E). Unexpectedly, the processivity of coexpressed L557P and K1050E telomerase was identical to that of K1050E, rather than intermediate between the 2 variants. In contrast, expression of K1050E with WT telomerase led to rescue of the processivity defect, and the degree of rescue was much greater than expected from a simple additive effect (Figure 3D-E). To confirm that the ability of L557P+K1050E telomerase to add many repeats to a DNA substrate was not simply additive between those of L557P and K1050E, Figure 3F shows band intensities in each lane of the gel. For the coexpression of WT and K1050E (top), one would expect the intensity of each band to be intermediate between those of WT and K1050E (given that there is half the amount of each enzyme) if the pattern of extension of WT+K1050E was simply a superimposition of those of WT and K1050E. However, for the longer products of extension (repeats ≥6; inset), the WT+K1050 (lavender) bands are almost as intense as WT (blue) for all products, whereas K1050E (peach) drops to baseline levels. Conversely, in the case of L557P+K1050E (bottom), the double mutant (green) follows exactly the same pattern as K1050E (peach), rather than being intermediate between it and L557P (red). This result indicates that L557P telomerase loses the ability to add >6 repeats to the substrate when expressed with K1050E, whereas the WT enzyme does not.
These data demonstrate that WT telomerase can rescue a processivity defect imparted by K1050E, whereas L557P telomerase cannot, explaining the much more severe phenotype of the proband relative to his heterozygous WT/K1050E mother.
L557P causes a defect in assembly of hTERT and hTR, whereas K1050E affects DNA substrate binding
To measure the ability of hTERT to bind stably to hTR, we quantitated relative recovery of hTR from an hTERT IP (Figure 4A), normalized to amount of recovery of hTERT protein (Figure 3A). As expected from our molecular modeling, the L557P variant resulted in a significant reduction in the ability of hTERT to bind to hTR, whereas K1050E had no effect (Figure 4B). The amount of reduction in RNA binding caused by L557P was the same as the amount of reduction in activity resulting from this mutation (Figure 3C), indicating that the specific activity (ie, activity per molecule of enzyme) was unchanged compared with WT. Thus, the only enzyme property that appears to be affected by L557P is affinity between hTERT and hTR.
![L557P causes a defect in telomerase assembly, whereas K1050E affects binding of telomerase to its DNA substrate. (A) Northern dot-blot showing the amount of hTR recovered after IP of hTERT from lysates of HEK293T cells expressing the indicated hTERT variants. Samples were normalized to the recovery of hTERT on western blots and loaded in duplicate, with their average used for quantitation. (B) Quantitation of the amount of hTERT-hTR binding from panel A; data are the mean ± standard error of the mean (SEM; n = 4). *P = .0193, by repeated measures 1-way analysis of variance (ANOVA), followed by Dunnett’s post hoc test. (C) DNA binding assay to determine primer affinity (KD) of telomerase variants. Purified human telomerase was incubated with a biotinylated DNA oligonucleotide primer (biotin-TTAGGG)3) at the indicated primer concentrations, and the amount of telomerase remaining in the supernatant after recovery of the DNA on NeutrAvidin beads was quantitated by northern dot-blot for hTR; more telomerase was bound at higher DNA concentrations and removed from the solution, leaving less free telomerase in the supernatant. (D) Quantitation of telomerase primer KD for the indicated hTERT variants. Plots of the amount of telomerase removed from the solution were fitted to the equation y = (Bmax[S])/(KD + [S]), where Bmax is the maximum level of binding, [S] is the concentration of DNA, and KD is the equilibrium binding constant. Data are the mean ± SEM (n = 3-7); ***P = .001, by ordinary 1-way ANOVA, followed by Dunnett’s post hoc test.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/12/10.1182_bloodadvances.2022007029/1/m_advancesadv2022007029f4.png?Expires=1769375554&Signature=cCodAcCZ6UofIAA3mu593Uvw5rqAS6kjuWrGwr~-0J7Zr21aWa4dBA0pSt5ceitE1j1dIUWK8LKfLDZlx8S4E9JHdqmE1lBtJ8wXiH8nzNlr736JJGQPnXFu55D-vohJLfaz5ssAmsbfIx6e7qFC2Alz~5n5bZvuak8Sp9KtSffxDhNxAgqsJJgkyTJ2MosRfISz4hd~WK2-DAEeCZD9LTbGXxBMupS7-WdgZmJJudPxaPeA6uzGGDeF0AzduSoa4H0XUv~X2Xl6qSimyzWZZtL2fUVNVPI6UAlM7lyB2ZP1SchxCmKjSZszPsv1hMoIQnZId0N3JhTMCMuz2TE~1g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
L557P causes a defect in telomerase assembly, whereas K1050E affects binding of telomerase to its DNA substrate. (A) Northern dot-blot showing the amount of hTR recovered after IP of hTERT from lysates of HEK293T cells expressing the indicated hTERT variants. Samples were normalized to the recovery of hTERT on western blots and loaded in duplicate, with their average used for quantitation. (B) Quantitation of the amount of hTERT-hTR binding from panel A; data are the mean ± standard error of the mean (SEM; n = 4). *P = .0193, by repeated measures 1-way analysis of variance (ANOVA), followed by Dunnett’s post hoc test. (C) DNA binding assay to determine primer affinity (KD) of telomerase variants. Purified human telomerase was incubated with a biotinylated DNA oligonucleotide primer (biotin-TTAGGG)3) at the indicated primer concentrations, and the amount of telomerase remaining in the supernatant after recovery of the DNA on NeutrAvidin beads was quantitated by northern dot-blot for hTR; more telomerase was bound at higher DNA concentrations and removed from the solution, leaving less free telomerase in the supernatant. (D) Quantitation of telomerase primer KD for the indicated hTERT variants. Plots of the amount of telomerase removed from the solution were fitted to the equation y = (Bmax[S])/(KD + [S]), where Bmax is the maximum level of binding, [S] is the concentration of DNA, and KD is the equilibrium binding constant. Data are the mean ± SEM (n = 3-7); ***P = .001, by ordinary 1-way ANOVA, followed by Dunnett’s post hoc test.
L557P causes a defect in telomerase assembly, whereas K1050E affects binding of telomerase to its DNA substrate. (A) Northern dot-blot showing the amount of hTR recovered after IP of hTERT from lysates of HEK293T cells expressing the indicated hTERT variants. Samples were normalized to the recovery of hTERT on western blots and loaded in duplicate, with their average used for quantitation. (B) Quantitation of the amount of hTERT-hTR binding from panel A; data are the mean ± standard error of the mean (SEM; n = 4). *P = .0193, by repeated measures 1-way analysis of variance (ANOVA), followed by Dunnett’s post hoc test. (C) DNA binding assay to determine primer affinity (KD) of telomerase variants. Purified human telomerase was incubated with a biotinylated DNA oligonucleotide primer (biotin-TTAGGG)3) at the indicated primer concentrations, and the amount of telomerase remaining in the supernatant after recovery of the DNA on NeutrAvidin beads was quantitated by northern dot-blot for hTR; more telomerase was bound at higher DNA concentrations and removed from the solution, leaving less free telomerase in the supernatant. (D) Quantitation of telomerase primer KD for the indicated hTERT variants. Plots of the amount of telomerase removed from the solution were fitted to the equation y = (Bmax[S])/(KD + [S]), where Bmax is the maximum level of binding, [S] is the concentration of DNA, and KD is the equilibrium binding constant. Data are the mean ± SEM (n = 3-7); ***P = .001, by ordinary 1-way ANOVA, followed by Dunnett’s post hoc test.
Telomerase processivity is affected by its affinity for its DNA substrate; lower DNA affinity leads to a greater probability of the enzyme dissociating after synthesis of each repeat.74,75 It has been reported that K1050E reduces the binding affinity of a C-terminal fragment of hTERT to a DNA oligonucleotide,37 and our molecular modeling supports this conclusion. To confirm this result with the full-length protein and measure DNA binding affinity of the combinations of variants, telomerase was incubated with a range of concentrations of a biotinylated telomeric DNA oligonucleotide, and the amount of telomerase recovered with the DNA on NeutrAvidin beads was quantitated61 (Figure 4C; supplemental Figure 2). K1050E resulted in an approximately eightfold increase in the equilibrium binding constant KD, whereas L557P had no effect (Figure 4D). Combining K1050E with either L557P or WT telomerase resulted in an approximately fourfold increase in KD compared with WT telomerase. Thus, in contrast to the differing effects of WT and L557P telomerase on the K1050E processivity defect, combining either enzyme with K1050E resulted in an intermediate level of DNA affinity.
Discussion
This study provides a thorough functional characterization of 2 missense variants in TERT, the gene encoding the catalytic subunit of telomerase, that are inherited in a compound heterozygous manner in an infant with the severe TBD Hoyeraal-Hreidarsson syndrome.46 Structural modeling predicted that each amino acid change would result in a different biochemical defect, with the novel variant L557P predicted to affect binding between hTERT and its RNA subunit, whereas K1050E would affect DNA binding and processivity. These predictions were borne out by biochemical analyses.
Together with the absence of either variant in population genome databases such as gnomAD, our functional data allow us to assign “likely pathogenic” status76 to both variants, even when each is present in a heterozygous state. K1050E has been reported in heterozygous form in 2 individuals with pulmonary fibrosis39,71 ; our functional analyses resolve a reported discrepancy in the effect of this variant on telomerase activity37,39 and confirm that it affects binding to the DNA-RNA duplex in the telomerase active site and results in a reduction in enzyme processivity.37 A different variant in hTERT that reduces telomerase processivity by ∼50% has been reported to be sufficient for telomere shortening in vivo and familial pulmonary fibrosis when inherited heterozygously.77 Therefore, although coexpression of K1050E and WT telomerase appeared to largely rescue the processivity defect of the mutant enzyme (Figure 3B), the likely pathogenicity of this heterozygous variant is supported in our study by the moderately short telomeres of the proband’s mother (Figure 1B-D), who is currently asymptomatic but will continue to be monitored for adult-onset pulmonary defects. Similarly, coexpression of L557P with WT telomerase appeared to rescue the effect of L557P on RNA binding and telomerase activity, which may contribute to the relatively long telomeres of the proband’s father. Nevertheless, the mild TBD features exhibited by the father and paternal grandmother suggest that this allele is mildly hypomorphic when heterozygous.
The most striking outcome of the functional analyses was the apparent functional interaction between the 2 missense hTERT variants when present in trans. Although coexpression of K1050E with WT telomerase rescued the processivity defect of the former allele, this was not the case when it was coexpressed with L557P. Indeed, the processivity of L557P+K1050E was identical to that of K1050E, rather than being intermediate between the 2 alleles (Figure 3E-F). The differing effects of WT or L557P hTERT on the processivity of their K1050E partner implies that RNA binding in one telomerase molecule can impact repeat addition processivity in the other molecule. The simplest explanation for this phenomenon would be an interaction between 2 catalytic subunits of a dimeric telomerase enzyme. There is evidence that the observed bilobal conformation of human telomerase is a result of the association of 2 hTERT molecules, each containing an active site,78 and other studies have provided biochemical evidence for functional interactions between 2 different telomerase variants.79-81 Nevertheless, a recent cryo-EM structural study interpreted the bilobal shape as containing only 1 hTERT molecule,38 and it therefore remains possible that the functional interaction we observed is a result of transient interactions of monomeric telomerase enzymes.
This study emphasizes the importance of thorough functional characterization of telomerase variants in addition to telomere length measurement in patients, to support definitive molecular diagnosis for families carrying such variants, which is necessary for treatment and reproductive decisions across the extended family. Such a diagnosis is particularly necessary for compound heterozygous variants, because the combined effects of 2 different alleles on telomerase function are impossible to predict.
Acknowledgments
Work in the Rare Disease Functional Genomics laboratory was supported by Luminesce Alliance - Innovation for Children’s Health, a not-for-profit cooperative joint venture between the Sydney Children’s Hospitals Network, Children’s Medical Research Institute, the Children’s Cancer Institute, the University of Sydney and the University of New South Wales, established with the support of the New South Wales government. Work in the Bryan laboratory was supported by generous donations from the Hill Foundation and J. and A. Smith. S.B.C. was supported by the Ernest and Piroska Major Foundation. H.A.P. was supported by National Health and Medical Research Council of Australia grant 1162886. Genomic characterization was performed as part of the Melbourne Genomics Health Alliance Bone Marrow Failure Flagship. L.C.F. is supported by a doctoral degree scholarship from Maddie Riewoldt’s Vision. Phosphorimaging analysis was performed on a Typhoon Biomolecular Imager purchased with support from the Ramaciotti Foundation.
Authorship
Contribution: A.N., A.M., S.B.C., L.G.R., and T.M.B. performed and analyzed all in vitro functional analyses of telomerase; J.T. and J.K.H. performed the molecular modeling; L.C.F. and P.B. performed and analyzed genomic characterization of the proband and family members; R.S.V., H.A.P., J.A.C., and P.M.B. performed the telomere length analyses; J. Roy and J. Rodgers collected clinical data and obtained informed consents; T.M.B. and A.N. wrote the manuscript with input from all authors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Tracy M. Bryan, Children’s Medical Research Institute, 214 Hawkesbury Rd, Westmead, NSW 2145, Australia; e-mail: tbryan@cmri.org.au.