Key Points
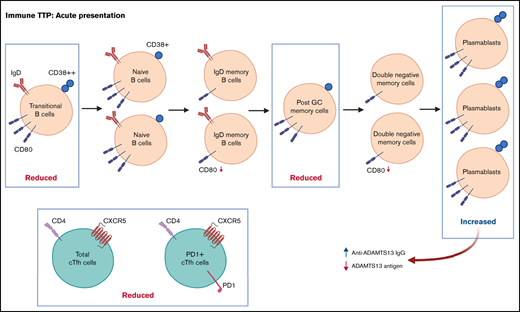
Abnormal B-cell phenotype in acute iTTP with decreased transitional and post–germinal center memory cells and increased plasmablasts.
Decreased total and PD1+ circulating T-follicular helper cells and changes in B-cell CD80 expression suggest altered B- and T-cell interactions.

Abstract
T follicular helper (Tfh) cells regulate development of antigen-specific B-cell immunity. We prospectively investigated B-cell and circulating Tfh (cTfh) cell subsets in 45 patients with immune thrombotic thrombocytopenic purpura (iTTP) at presentation and longitudinally after rituximab (RTX). B-cell phenotype was altered at acute iTTP presentation with decreased transitional cells and post–germinal center (post-GC) memory B cells and increased plasmablasts compared with healthy controls. A higher percentage of plasmablasts was associated with higher anti-ADAMTS13 IgG and lower ADAMTS13 antigen levels. In asymptomatic patients with ADAMTS13 relapse, there were increased naïve B cells and a global decrease in memory subsets, with a trend to increased plasmablasts. Total circulating Tfh (CD4+CXCR5+) and PD1+ Tfh cells were decreased at iTTP presentation. CD80 expression was decreased on IgD+ memory cells and double-negative memory cells in acute iTTP. At repopulation after B-cell depletion in de novo iTTP, post-GC and double-negative memory B cells were reduced compared with pre-RTX. RTX did not cause alteration in cTfh cell frequency. The subsequent kinetics of naïve, transitional, memory B cells and plasmablasts did not differ significantly between patients who went on to relapse vs those who remained in remission. In summary, acute iTTP is characterized by dysregulation of B- and cTfh cell homeostasis with depletion of post-GC memory cells and cTfh cells and increased plasmablasts. Changes in CD80 expression on B cells further suggest altered interactions with T cells.
Introduction
Immune thrombotic thrombocytopenic purpura (iTTP) is a life-threatening thrombotic microangiopathy mediated by an immunoglobulin G (IgG) antibody against the metalloprotease ADAMTS13 that enhances its clearance or inhibits its von Willebrand factor–processing activity.1 In iTTP, there is an incompletely understood loss of tolerance resulting in a shift from immune homeostasis to autoimmunity. This involves dendritic cells, which acquire antigens derived from ADAMTS13 that activate cross-reactive naïve CD4+ T cells, which differentiate into autoreactive effector CD4+ T cells.2,3 Mature autoreactive B cells recirculate into the germinal center (GC) of secondary lymph nodes, where they are stimulated by compatible antigens in the presence of the autoreactive T helper cells and differentiate into autoantibody-producing plasma cells or long-lived memory B cells.4
B-cell depletion therapy with rituximab (RTX; a chimeric monoclonal antibody against the pan–B-cell marker CD20) has been demonstrated to be effective in reducing relapse rates in iTTP and prolonging disease-free survival in acute episodes, compared with PEX and steroid alone.5-7 Giving RTX preemptively in patients at high risk of a clinical relapse (based on a fall in ADAMTS13 activity levels to <10% to 20%; ie, ADAMTS13 relapse) reduces clinical relapse rates compared with historical controls.8
A recent Genome Wide Association Study (GWAS) of iTTP confirmed associations with single nucleotide polymorphisms (SNPs) at the HLA locus and identified a novel association on chromosome 3.9 The locus on chromosome 3 contains 5 genes, 1 of which is the CD80 gene. CD80 is a costimulator for T lymphocyte activation. After activation through the B-cell receptor or interleukin-4 (IL-4), B cells express CD80, which interacts with CD28 on T cells to provide costimulation signals.10 The HLA-DRB1*11 and DRB1*03 genes are known risk factors for iTTP, suggesting the major histocompatibility complex class II protein variants they encode have optimal affinity for certain ADAMTS13 peptides recognized by CD4+ T-cell receptors in patients with iTTP.11,12 Two peptides derived from the CUB-2 domain of ADAMTS13 are presented on HLA-DRB1*11 and HLA-DRB1*03, respectively, and are recognized by iTTP patient-derived CD4+ T cells.13,14
CD4+ T cells are pivotal in development of iTTP because CD4+ T-cell help is required in the production and affinity maturation of ADAMTS13-directed antibodies.3 Within the CD4+ T-cell population, the T follicular helper (Tfh) subset is vital for supporting antibody-mediated immune responses by providing costimulation signals through CD40L and IL-21 production, which promotes the growth, differentiation, and class-switching of antigen-activated naïve B cells.15-17 Tfh cells constitutively express the chemokine receptor CXCR5, which facilitates migration into GCs.18 Tfh cells express costimulatory molecules such as inducible costimulatory molecule (ICOS) and immune-regulatory molecules such as programmed death-1 (PD-1) as their transcription factors, which can be used to further define Tfh cells.19 More specifically, PD−1+ICOS− Tfh cells seem to represent quiescent and PD−1+ICOS+ Tfh cells recently activated memory Tfh cells.20,21
Circulating Tfh (cTfh) cells also express the GC homing receptor CCR7.22 Expansion of cTfh cells has been associated with the development of several autoimmune diseases.23-29
The aim of this study was to investigate the B-cell and cTfh subset distribution in patients with iTTP, both at presentation and longitudinally after anti-CD20 therapy in relation to clinical and laboratory parameters and B-cell kinetics. The role of cTfh cells has not previously been investigated in iTTP. The temporal relationships between B- and T-cell subpopulation at critical stages throughout the course of disease will improve the understanding of the pathogenesis of iTTP, potentially provide biomarkers to predict relapse, and potentially identify new avenues for therapeutic intervention.
Methods
Patients and controls
We prospectively enrolled patients with iTTP from September 2018 to June 2021 through the United Kingdom TTP Registry (database and Biobank of UK TTP–Multicentre Research Ethics Committee [MREC] :08/H0810/54 and MREC:08/H0716/72). The study was conducted according to the Declaration of Helsinki. Blood samples were obtained from age- and sex-matched healthy controls (HCs) with no history of autoimmune disease or immunosuppressant medication. Definitions of iTTP diagnosis, remission, and relapse were based on previous studies.30-32
Patients were divided into 2 groups: those with de novo acute iTTP episodes and asymptomatic ADAMTS13 relapses (treated with preemptive RTX). Acute presentations of iTTP were treated with PEX, corticosteroids, and RTX (4-8 doses, 375 mg/m2 to normalize ADAMTS13 activity). Time after RTX was defined as time since first RTX infusion. Caplacizumab was given to 18/22 patients.
Patients with previously diagnosed iTTP who were in clinical remission (normal platelet count) but developed a new severe ADAMTS13 deficiency were referred to as ADAMTS13 relapse episodes.32 These patients received preemptive RTX (4 doses of 200/500/375 mg/m2 1 week apart). Dosing varied at clinician discretion or as part of a separate randomized control trial (Elective Rituximab in TTP trial; [REC]17/LO/1055).
Blood samples and PBMC isolation
Heparinized blood samples collected from patients prior to initiation of RTX or other immunosuppression and HC were analyzed within 24 hours or frozen at −80°C and stored for later analysis. Samples were also collected 1 month and 3 months after RTX and then every 3 months thereafter until clinical or ADAMTS13 relapse or end of study (whichever came first). Peripheral blood mononuclear cells (PBMCs) were isolated by diluting blood (1:1) with phosphate-buffered saline 1× and layered over Ficoll-Paque Premium 1084R (Sigma-Aldrich).33
Flow cytometry
PBMCs (approximately 1 × 106/sample) were incubated with conjugated antibodies for 20 minutes in the dark at room temperature. Cells were washed twice and resuspended in phosphate-buffered saline, and samples were analyzed on a Cytoflex S cytometer (Beckman Coulter) (supplemental Materials). Each sample was divided into 2 antibody panels: for B-cell subsets and Tfh cells (supplemental Materials, Table 1A-B). Viability was determined using LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit (Invitrogen). Analysis was performed on FlowJo software version 10 (FlowJo LLC).
B-cell immunophenotyping and gating strategy
We used the mature B cell Bm1–Bm5 classification to identify B-cell subsets based on coexpression of IgD and CD38. B-cell subsets include transitional B cells, naïve B cells, memory populations (including IgD+ memory cells), post-GC cells, double-negative memory cells, and plasmablasts.34,35 Antibodies used for the B-cell panel were CD19 PECy7, CD27 APC, IgD BV421, and CD38 PE. Following from our GWAS finding that the CD80/POGLUT1 locus is associated with iTTP,9 we also analyzed the level of CD80 expression (median fluorescence intensity [MFI]) on B-cell subsets in iTTP.36 Gating strategies are shown in supplemental Figure 1.
Circulating Tfh cell immunophenotyping and gating strategy
The total cTfh cell population was defined by CD3+ T cells that expressed CD4+CXCR5+. The different subsets within the total cTfh cell population were defined by expression of activation markers: PD1+ cTfh, ICOS+ cTfh, and PD1+ICOS+ cTfh. In the cTfh panel, PBMCs were stained with CD3 PECy7, CD4 PerCP, CXCR5 FITC, PD1 APC, and ICOS BV421 antibodies. The gating strategy and representative plots are shown in supplemental Figure 2.
ADAMTS13 assays
ADAMTS13 activity was analyzed by fluorescence resonance energy transfer (normal range, 60% to 123%).37 ADAMTS13 antigen levels were quantified using in-house–developed, enzyme-linked immunosorbent assay, previously described (normal range, 74% to 134%).38 ADAMTS13 activity and antigen levels were expressed as a percentage relative to pooled normal plasma. Anti-ADAMTS13 IgG levels were measured with in-house enzyme-linked immunosorbent assay (normal range <6.1%) with concentration of anti-ADAMTS13 IgG calculated as a percentage relative to an index plasma from a patient with a high auto-antibody titer (assigned a value of 100%).7
Statistical analysis
Mann-Whitney U and Wilcoxon tests were used for paired and unpaired continuous variables, respectively. The X2 and Fisher’s exact tests were performed for categorical variables. Spearman correlation test was used to measure the possible relationship between 2 variables of interest. Statistical analysis was performed using GraphPad Prism 8 (GraphPad Software Inc.).
Results
Patient demographics and ADAMTS13 biomarkers
Demographics of patients with iTTP and HC and ADAMTS13 assay results are shown in supplemental Table 2A. There were 45 unique patients with iTTP involving 46 episodes: 22 de novo acute episodes and 24 asymptomatic ADAMTS13 relapses treated with preemptive RTX.
In acute iTTP episodes, 82% (18/22) of cases had cardiac involvement and 50% (11/22) had neurological involvement. In 14% (3/22) of acute episodes, patients required organ support in the intensive care unit. All acute episodes were treated with PEX, corticosteroids, and RTX; 82% (18/22) of patients received anti–von Willebrand factor nanobody, caplacizumab. Additional immunosuppression included mycophenolate mofetil (27%, 6/22) and bortezomib (9%, 2/22). All but 1 patient with ADAMTS13 relapse episodes in this study had previously received RTX treatment, during either an acute presentation or previous ADAMTS13 relapse episode. In this cohort, the median number of previous TTP episodes was 3 (range, 1-8), with a median duration since the most recent TTP episode being 21 months (range, 13-191) (supplemental Table 2B).
Post-GC memory B cells are decreased and plasmablasts increased at iTTP presentation
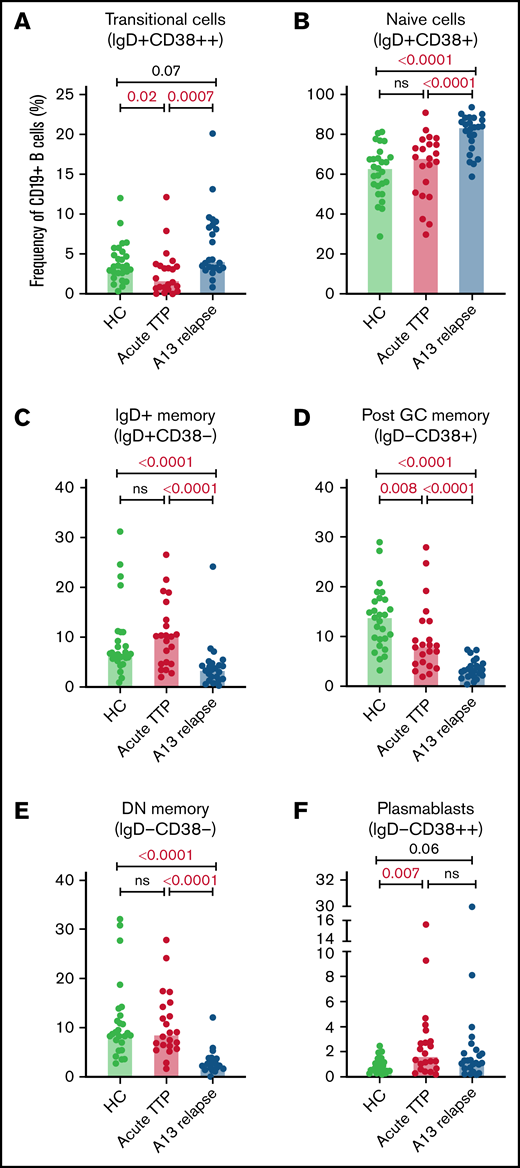
The total number of CD19+ B cells at presentation of acute iTTP or preceding preemptive RTX for ADAMTS13 relapse episodes was not significantly different than HC. At presentation in acute iTTP episodes, post-GC memory cells were decreased compared with HCs (13.7%; range, 3.1% to 28.8%) vs acute iTTP (7.9%; range, 1.8% to 27.8%; P = .008), whereas plasmablasts were increased (HC 0.7%; range, 0.2% to 2.5%) vs acute iTTP (1.6%; range, 0.2% to 15.4%; P = .007) (Figure 1).
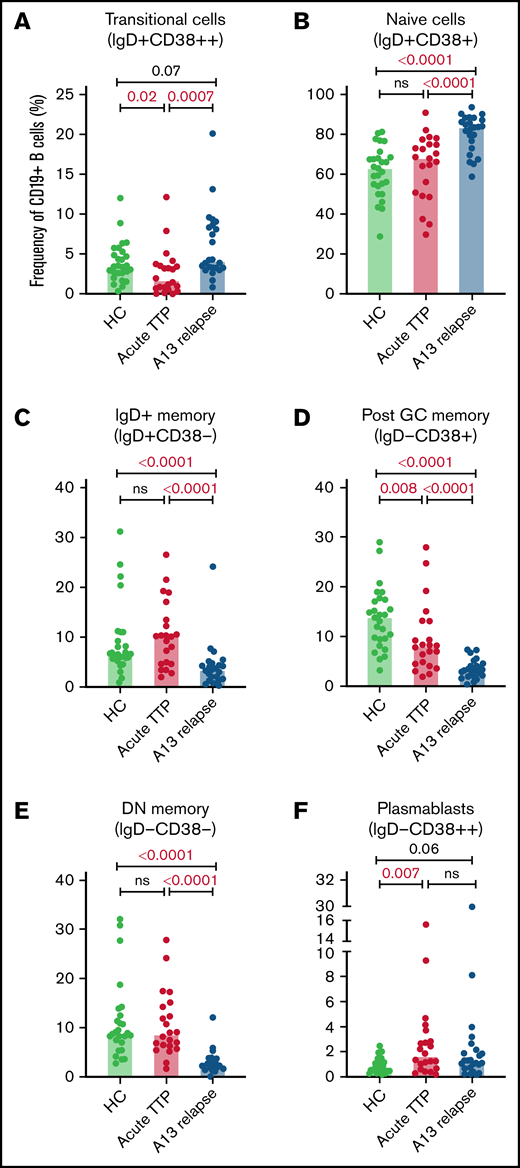
B cell subsets at iTTP presentation and ADAMTS13 relapse. Percentages of transitional cells (IgD+CD38++) (A), naïve cells (IgD+CD38+) (B), IgD+ memory B cells (IgD+CD38–) (C), postgerminal center memory B cells (IgD–CD38+) (D), double–negative memory B cells (IgD–CD38–) (E), and plasmablasts (IgD–CD38++) (F) in patients with acute iTTP episodes (n = 22), ADAMTS13 relapse (n = 24), and HC (n = 27). A13 relapse, ADAMTS13 relapse; double-negative memory cells, double-negative memory cells; Post GC memory, postgerminal center memory cells.
B cell subsets at iTTP presentation and ADAMTS13 relapse. Percentages of transitional cells (IgD+CD38++) (A), naïve cells (IgD+CD38+) (B), IgD+ memory B cells (IgD+CD38–) (C), postgerminal center memory B cells (IgD–CD38+) (D), double–negative memory B cells (IgD–CD38–) (E), and plasmablasts (IgD–CD38++) (F) in patients with acute iTTP episodes (n = 22), ADAMTS13 relapse (n = 24), and HC (n = 27). A13 relapse, ADAMTS13 relapse; double-negative memory cells, double-negative memory cells; Post GC memory, postgerminal center memory cells.
B-cell repopulation after RTX recapitulates ontogeny beginning with naïve B-cell exit from the bone marrow followed by gradual maturation of memory subsets over time. In ADAMTS13 relapse cases, there was an increased proportion of naïve B cells compared with HCs and a trend to increased transitional cells and plasmablasts (Figure 1). There was a marked reduction in all memory subsets.
Altered circulating Tfh cell subsets at iTTP presentation: decreased total cTfh and PD1+ cTfh cells
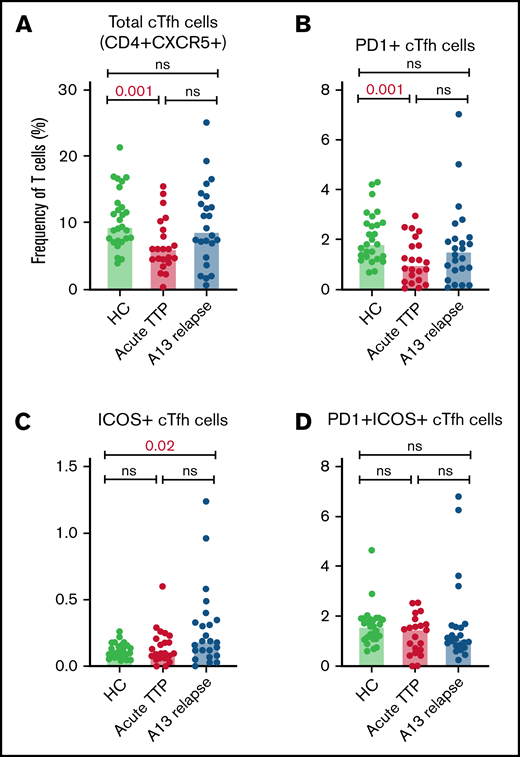
The relative proportions of CD4+ cTfh subsets in acute TTP and ADAMTS13 relapse were determined and compared with HCs (Figure 2). The frequency of total cTfh and PD1+ cTfh was significantly reduced in patients with acute iTTP compared with HCs. This may be suggestive of migration of circulating Tfh cells into GCs. There were no significant differences seen in ICOS+ or PD1+ICOS+ cTfh cells. In ADAMTS13 relapses, no differences were seen in total, PD1+, or PD1+ICOS+ cTfh cells. However, ICOS+ cTfh cells were increased in patients compared with HCs.
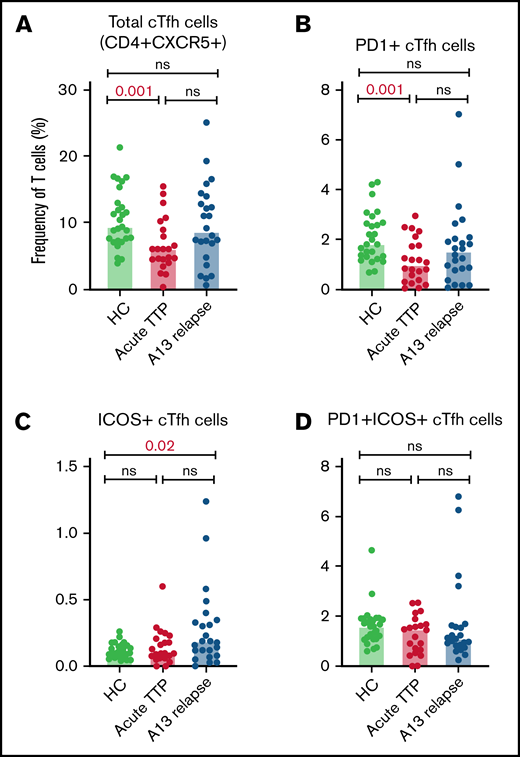
Circulating Tfh subsets at iTTP presentation and ADAMTS13 relapse. Percentages of total cTfh (CD4+CXCR5+) (A), PD1+ cTfh (CD4+CXCR5+PD1+) (B), ICOS+ cTfh (CD4+CXCR5+ICOS+) (C), and PD1+ICOS+ cTfh (CD4+CXCR5+PD1+ICOS+) (D) in patients with acute iTTP episodes (n = 34), ADAMTS13 relapse (n = 27), and HC (n = 27).
Circulating Tfh subsets at iTTP presentation and ADAMTS13 relapse. Percentages of total cTfh (CD4+CXCR5+) (A), PD1+ cTfh (CD4+CXCR5+PD1+) (B), ICOS+ cTfh (CD4+CXCR5+ICOS+) (C), and PD1+ICOS+ cTfh (CD4+CXCR5+PD1+ICOS+) (D) in patients with acute iTTP episodes (n = 34), ADAMTS13 relapse (n = 27), and HC (n = 27).
Relationship among ADAMTS13 parameters, circulating Tfh cells, and B-cell subsets at iTTP presentation
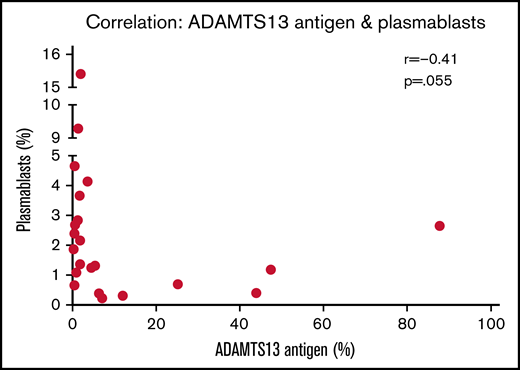
In the acute iTTP group, median time to achieve a normal platelet count from presentation was 4 days (range, 2-11) and median time to normalization of ADAMTS13 activity was 33 days (range, 3-382; interquartile range, 22-124). At presentation in acute iTTP episodes, a higher percentage of plasmablasts appears to be associated with lower antigen levels (r = −0.41; P = .055) (Figure 3). Indeed, a plasmablast level of >3% was associated with IgG antibody level of >50% (Table 1). This may be due to increased production of anti-ADAMTS13 IgG antibody from a larger number of plasmablasts, which in turn results in increased ADAMTS13 clearance. In both acute iTTP cases and ADAMTS13 relapses, there was no correlation between total cTfh cells and any of the B-cell subsets (data not shown).
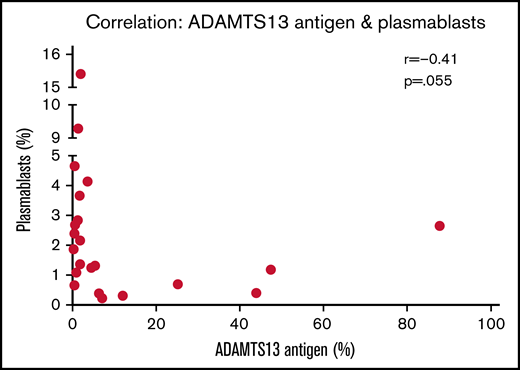
Relationship between ADAMTS13 antigen levels and plasmablast frequency. Spearman correlation analysis was performed, and P < .05 indicates that the difference is statistically significant.
Relationship between ADAMTS13 antigen levels and plasmablast frequency. Spearman correlation analysis was performed, and P < .05 indicates that the difference is statistically significant.
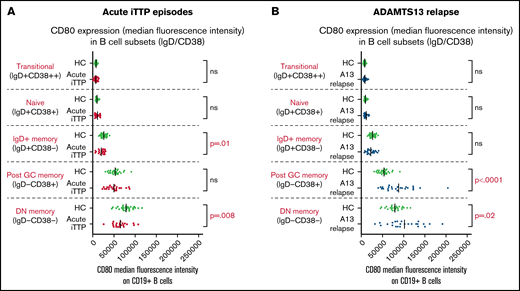
Expression of CD80 on B-cell subsets at acute presentation and after RTX
In acute iTTP episodes, CD80 MFI was decreased in IgD+ memory cells and double-negative memory cells compared with HCs (Figure 4). However, in ADAMTS13 relapse cases, CD80 MFI was significantly increased in post-GC and double-negative memory cells compared with HCs. A possible explanation for this difference between acute iTTP and asymptomatic ADAMTS13 relapse may be that in asymptomatic falls in ADAMTS13 activity prior to an acute clinical relapse, it is possible to detect activated memory B-cell populations due to less background noise or before they marginate.
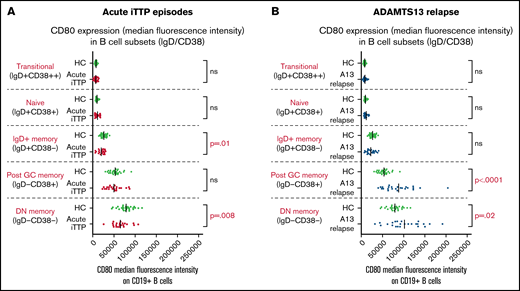
Analysis of CD80 expression (median fluorescence intensity) on B-cell subsets defined by IgD/CD38. Acute iTTP cases (A) and ADAMTS13 relapse cases (B).
Analysis of CD80 expression (median fluorescence intensity) on B-cell subsets defined by IgD/CD38. Acute iTTP cases (A) and ADAMTS13 relapse cases (B).
A summary of the B- and Tfh cell immunophenotyping results at acute iTTP presentation and ADAMTS13 relapse is shown in Table 2.
Longitudinal analysis: effect of RTX
Flow cytometric analysis of B-cell subsets and cTfh cells was performed for patients before RTX and longitudinally at 1 month, 3 months, 6 months, 9 months, 12 months (ie, every 3 months) until end of study/relapse. After RTX treatment, all 46 episodes achieved B-cell depletion (defined by a laboratory CD19+ count <0.005 × 109/L), except for 1 patient in the ADAMTS13 relapse cohort (supplemental Figure 3A-F for acute iTTP cases; data not shown for ADAMTS13 relapse episodes). In contrast, RTX did not cause any significant alterations in cTfh numbers (supplemental Figure 3G-K for acute iTTP cases; data not shown for ADAMTS13 relapse episodes). B-cell return occurred at median 10 months (range, 6-14) in the acute iTTP group and 8 months (range, 0.25-15) in ADAMTS13 relapse group. The timepoint of B-cell return was not associated with ADAMTS13 or clinical relapse.
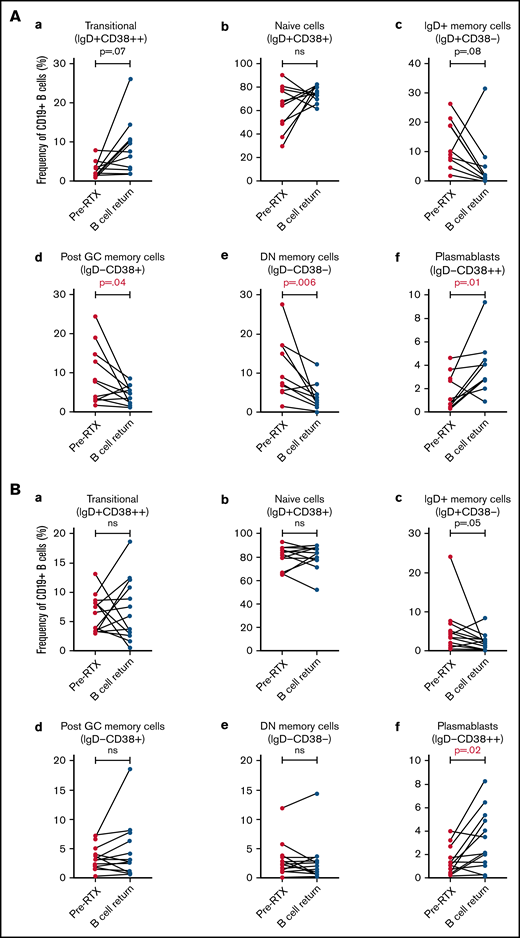
Comparison of B-cell subsets frequencies prior to RTX treatment and at B-cell return
We then compared 2 specific timepoints: before RTX and B-cell return. Repopulation following B-cell depletion in the acute iTTP group demonstrated relatively higher proportion of plasmablasts but a reduction in post-GC and double-negative memory B cells (Figure 5A). In the ADAMTS13 relapse group, in which all except 1 patient had received historical RTX, plasmablasts were increased in frequency at B-cell return compared with pretreatment levels, but the distribution of B-cell subsets was otherwise similar before and after retreatment (Figure 5B).
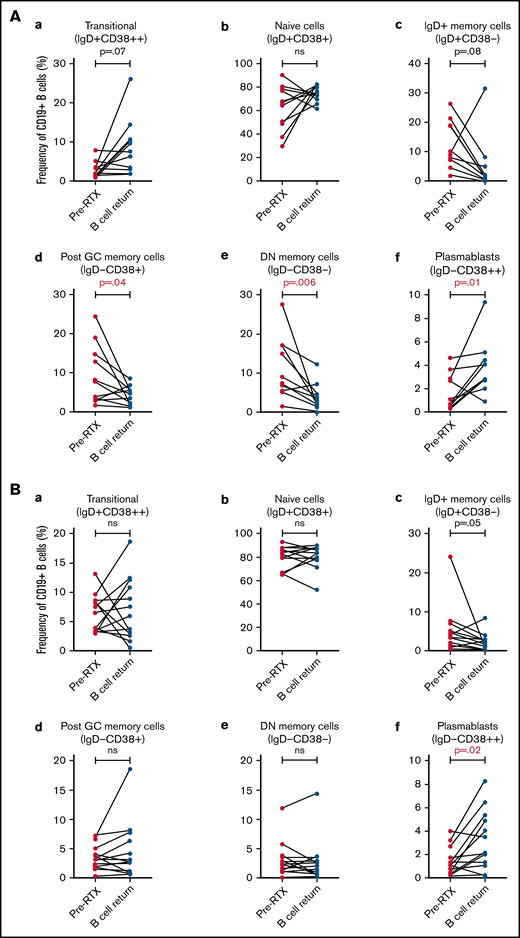
Pairwise comparison of B-cell subset frequencies before and after RTX by Wilcoxon signed–rank test. (A) Acute iTTP episodes, pairwise comparison of 10 patients. (B) ADAMTS13 relapse episodes, pairwise comparison of 13 patients.
Pairwise comparison of B-cell subset frequencies before and after RTX by Wilcoxon signed–rank test. (A) Acute iTTP episodes, pairwise comparison of 10 patients. (B) ADAMTS13 relapse episodes, pairwise comparison of 13 patients.
Longitudinal follow-up: relapses vs sustained remissions
We prospectively investigated changes in B-cell subsets after RTX in 20 patients with iTTP (16 from our initial cohort and 4 additional patients) longitudinally until a subsequent clinical or ADAMTS13 relapse and compared with patients who remained in remission over an equivalent period.
There were 10 patients in the relapse cohort (3 followed after an acute episode, 7 after elective RTX) and 10 in the remission cohort (4 acute and 6 elective episodes at t = 0). Median age was 51 years (range, 38-81) and 45.5 years (range, 21-68), respectively. Median follow-up was 15 months (range, 6-24). One patient in the relapse group did not achieve B-cell depletion (CD19+count 0.005 × 109/L). Time to B-cell return was similar: 8 months (range, 4-13) in relapses vs 8 months in remission (range, 0.25-15.5). All subsequent relapses were ADAMTS13 relapses and occurred at a median of 14 months (range, 9-25).
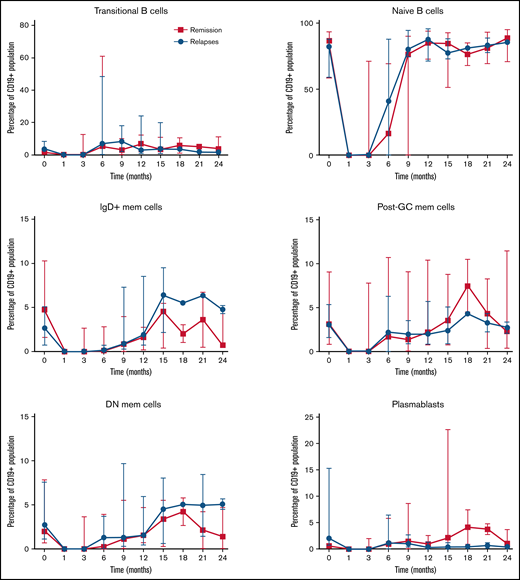
Longitudinal analysis showed that in both groups, B-cell return after RTX develops along normal B-cell ontogeny. The kinetics of naïve, transitional, memory B cells and plasmablasts did not differ significantly between patients who subsequently experienced relapse vs those who remained in sustained remission (Figure 6). No alterations in B-cell subsets were identified before a relapse.
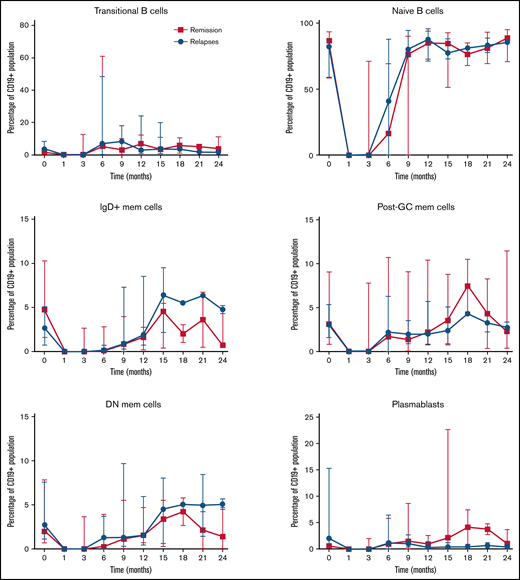
Longitudinal changes in B-cell subsets after RTX therapy for an acute iTTP episode or ADAMTS13 relapse (ADAMTS13 activity 15%) until subsequent ADAMTS13 relapse, compared with patients who remained in remission over an equivalent period.
Longitudinal changes in B-cell subsets after RTX therapy for an acute iTTP episode or ADAMTS13 relapse (ADAMTS13 activity 15%) until subsequent ADAMTS13 relapse, compared with patients who remained in remission over an equivalent period.
Discussion
Interactions between T and B cells that occur within GCs of secondary lymphoid organs are critical for the development of humoral immune responses. Tfh cells, a distinct subset of T helper cells, have been recognized in recent years as a crucial regulator of GC formation, B-cell development, and long-term humoral memory generation and play a significant role in the pathogenesis of autoimmune diseases.17 The mechanism involved in the loss of tolerance and subsequent development of anti-ADAMTS13 antibodies in patients with iTTP is still largely unknown, and we currently lack the ability to predict relapse accurately. The observed association between the major histocompatibility complex class II allele HLA DRB1*11 and development of iTTP implies a role for helper CD4+ T cells in the initiation of autoimmune reactivity against ADAMTS13.11,12,39
This is the first prospective study to perform a comprehensive analysis of B-cell subsets and cTfh cell changes in iTTP before and after immunosuppressive therapy. B-cell phenotype is altered at acute iTTP presentation with decreased post-GC memory B cells and an increase in plasmablasts in iTTP compared with HCs. Potential mechanisms for the reduced proportion of memory B cells among circulating B cells at presentation of iTTP include (1) some B cells dying in the early memory B-cell stages and never becoming memory B cells, (2) hyperactivation of T and B cells in iTTP resulting in an elevated differentiation of memory B cells into antibody-producing plasma cells, and (3) migration of memory B cells into lymphoid tissue.40-42 Similar reductions in the memory B-cell compartments have been described in sarcoidosis, systemic sclerosis, and Sjögren syndrome.40,43 ,, -48
Comparing immunophenotype results between different studies is complex due to the variety of staining and gating protocols used and clinical and demographic characteristics of the patient cohort, but some general conclusions can be made regarding iTTP in comparison to other autoimmune diseases. We found an increased ratio of naïve to switched memory B cells and higher plasmablasts in the acute patients. In contrast, in patients with active systemic lupus erythematosus (SLE) there is an apparent expansion of memory vs naïve B-cell subsets correlating in part with disease activity, but this is due to a naïve B-cell lymphopenia.49 Expansion of plasmablasts also occurs in SLE but is relatively higher than we found in iTTP. In Sjögren syndrome, transitional and naïve B cells seem expanded compared with memory subsets, thus closer to the iTTP result, but in patients with rheumatoid arthritis, naïve and memory populations are generally similar to healthy age-matched controls.50 More recently, attention has focused on the double-negative (IgD−CD27−) B-cell subset. In systemic sclerosis, SLE, and RA, the relative importance of this subset, especially in relation to therapy resistance, has been recognized. The double-negative B-cell population contains both activated and antibody-secreting post-GC B cells and very early switched B cells (with fewer mutated IgG transcripts).51,52 This population was not different from HCs in the patients with iTTP in relation to relapse, but additional studies would be needed to assess functional properties of B cells within this population in iTTP.
The relative contribution of different naïve and antigen-experienced B-cell subsets to pathogenesis and response to therapies will depend on the position and circumstances underlying specific breaches of tolerance and thus ultimately depend on the specificity of the autoreactive B-cell receptor(s). The different patterns we have described in iTTP compared with other systemic autoimmune diseases indicate a lack of apparent expansion of memory populations in the peripheral blood and thus the probability of rapid sequestration or expansion of autoantibody-producing clones within tissues or lymphoid organs.
At acute iTTP presentation, we also demonstrate a novel association between higher plasmablast frequency and higher ADAMTS13 IgG antibody level and a trend toward reduced ADAMTS13 antigen levels. This suggests a potential underlying mechanism for iTTP development, in which rapid differentiation to autoantibody-producing plasmablasts leads to increased production of anti-ADAMTS13 IgG, which in turn results in increased ADAMTS13 clearance.
In asymptomatic patients with ADAMTS13 relapse, alterations in B-cell subsets prior to preemptive RTX therapy were pronounced with significantly increased naïve B-cell population, global decrease in all memory subsets, and trend toward increased plasmablasts. These subset alterations most likely represent changes related to historic RTX therapy, with long-term suppressive effects of RTX on memory cell subsets. This also explains the largely similar distribution of B-cell subsets before and after preemptive RTX treatment. After B-cell depletion therapy, B cells repopulate primarily from bone marrow–derived naïve B cells, with delayed regeneration of memory B cells.53,54 Poor memory B-cell reconstitution may reflect long-term effects of RTX on B cells, described after organ transplantation and in autoimmune diseases.55-57
Total cTfh (CD4+CXCR5+) and PD1+ Tfh cells were decreased in acute patients with iTTP at presentation compared with HC. A similar decrease in cTfh cells has been observed in sarcoidosis with infiltration of Tfh cells into skin lesions, suggesting that cTfh cells are recruited into affected sites.47 In acute iTTP, a potential explanation for the concomitant decrease in post-GC memory cells and cTfh cells may be that they are localizing in GCs within lymphoid tissue. In contrast, in asymptomatic patients with ADAMTS13 relapse, there were no significant differences in total cTfh cells and PD1+ cTfh cells. This may be because this is a different stage of the disease process or possibly because the effect of previous RTX alters the interplay of B and cTfh cells. Tfh T cells use CXCR5 positivity to access GCs. In GCs, they produce IL-21, which plays a key role in class switch and affinity maturation, leading the generation of memory B cells and plasma cells from antigen-activated naïve B cells. Other peripheral blood helper T cells (CXCR5−, CXCR2, and PD1+) can also produce IL-21 and have been identified as a source of extrafollicular T cell helper to B cells in patients with other autoimmune diseases.58 Although there are no frank inflammatory sites or lymphoid clusters in iTTP, it is possible that aberrant interactions between ADAMTS13-specific memory B cells and other T helper cells may occur in extrafollicular sites (eg, spleen).
We also compared B and cTfh frequencies at 2 different timepoints after RTX infusion: before RTX and at B-cell return. B-cell depletion was achieved in all patients by 1 month. Subsequent B-cell reconstitution occurred at 10 months in the acute iTTP cohort and 8 months in the asymptomatic ADAMTS13 relapse cohort. At B-cell return after a de novo acute iTTP episode, plasmablast levels were significantly raised, with a trend toward increased transitional and naïve cells. Importantly, no patients relapsed at the time of B-cell return. The time interval between B-cell return and relapse suggests that additional differentiation and selection of specific autoreactive B-cell clones from naïve populations may play a key role, although expansion of ADAMTS13 specific memory B-cell populations in lymphoid tissues to critical levels cannot be ruled out. RTX did not cause any significant alterations in cTfh cell frequency.
Our data also reveal altered expression of CD80 on B cells in iTTP, with decreased expression in IgD+ memory cells and double-negative memory cells in acute iTTP episodes and increased expression in post-GC memory and double-negative memory cells in ADAMTS13 relapses, prior to RTX therapy. CD80 and CD86 are expressed on antigen-presenting cells, such as dendritic cells and activated B cells, and have the capacity to stimulate or inhibit T-cell responses through their receptors CD28 and cytotoxic T lymphocyte-associated antigen-4 (CTLA-4), respectively.10 Dendritic cells exposed to ADAMTS13 have previously been shown to present ADAMTS13 peptides, with preferential HLA-DRB1* 11–dependent presentation of CUB2-derived peptides, which the authors hypothesized may contribute to the onset of acquired TTP by stimulating low-affinity, self-reactive CD4+ T cells.2,3
Manipulation of the CD80/CD86 pathway has shown efficacy in the clinical setting, with abatacept (a CTLA-4 immunoglobulin that targets autoimmune B cells by reducing CD80/CD86 expression) approved for the treatment of rheumatoid arthritis.59 Of interest, the CD80 locus was identified in the GWAS for iTTP and contained an SNP in the 3′ untranslated region that was in high linkage with the lead SNP.9 Further investigation into the link between possession of this SNP with translation and expression of CD80 on different cell types is ongoing. Additional studies are required to understand how these differences in expression could translate into functional properties of CD80/86 in patients with iTTP.
In the longitudinal analysis, no alterations in B-cell subsets were identified before a relapse, suggesting that temporal relationships between B-cell subpopulations cannot be used as biomarkers to better predict relapses.
Taken together, our findings give a novel insight into the role of B and cTfh cells in the development of iTTP. We propose that de novo acute iTTP is characterized by dysregulation of B- and cTfh cell homeostasis with decreased circulating subsets of GC memory cells and cTfh cells and an increased frequency of plasmablasts in the circulation, perhaps reflecting the increase in cognate interaction between antigen-specific T- and B-cell populations in secondary lymphoid tissue. Changes in the frequency of CD80 on B cells suggest altered interactions with T cells. The finding of alteration in CD80 expression further supports the recent novel association between iTTP and 5 alleles within a haploblock on chromosome 3, 1 of which encodes CD80.9 Although our studies did not address the questions of antigen selectivity and exquisite specificity of the autoimmune response to ADAMTS13 in iTTP, the novel findings will direct further functional analysis and provide potentially interesting targets for further research and therapeutics in iTTP.
Authorship
Contribution: J.-S.S. and M.O.S. designed research, recruited patients, performed laboratory testing, collected data, analyzed data, and wrote the manuscript; G.C. and R.d.G. designed research, analyzed data, and wrote the manuscript; Y.G. performed laboratory testing, collected data, and reviewed the manuscript; and M.S. and M.T. designed research, recruited patients, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: M.S. received speaker’s fees and honoraria from Alexion, Sanofi, Novartis, and Takeda, and has received research funding from Takeda. M.T. received speaker’s fees and honoraria from Sanofi and Bayer. The remaining authors declare no competing financial interests.
Correspondence: Mari Thomas, Department of Haematology, University College London Hospitals, 250 Euston Rd, London, United Kingdom, NW1 2PG; e-mail: mari.thomas@nhs.net.