

Key Points
This is the first fatal TMA following SMN AAV9 gene therapy in a child carrying a potential predisposing risk factor in the complement factor I gene.
A better understanding of the pathophysiology underlying TMA induced by AAV gene therapy is essential to ensure patient safety.
Abstract
Adeno-associated virus (AAV) gene therapies are highly promising, such as the onasemnogene abeparvovec (Zolgensma) in spinal muscle atrophy (SMA). We report the first case of fatal systemic thrombotic microangiopathy (TMA) following onasemnogene abeparvovec in a 6-month-old child with SMA type 1, carrying a potential genetic predisposition in the complement factor I gene. Other cases of TMA have recently been reported after onasemnogene abeparvovec and after AAV9 minidystrophin therapy in Duchenne muscular dystrophy. The risk-benefit ratio of this therapy must therefore be assessed. Early recognition of TMA and targeted immunotherapy are fundamental to ensure the safety of patients treated with AAV gene therapies.
Introduction
Spinal muscular atrophy (SMA) linked to chromosome 5q is a recessive genetic disease characterized by muscle weakness and atrophy, resulting in progressive degeneration and irreversible loss of α motor neurons in the spinal cord. Infantile-onset SMA, or SMA type 1, is typically characterized by decreased motor skills before 6 months of age, progressive respiratory failure, and premature death before the age of 2 years, despite supportive care.1 Onasemnogene abeparvovec (Zolgensma, Novartis, Basel, Switzerland), the adeno-associated virus (AAV) SMN gene therapy, has shown promising results in terms of efficacy in reducing premature death and safety in SMA.2 Thus far, more than 1400 patients have been treated with this gene therapy; however, there is still limited information on its possible harmful side effects.3-6 We report a fatal case of systemic thrombotic microangiopathy (TMA) following onasemnogene abeparvovec in a 6-month-old child with SMA type 1, emphasizing the need for close monitoring and targeted immunotherapy to optimize early TMA recognition and treatment by AAV gene therapy.
Methods
A 3-month-old girl, born to first cousins of Moroccan origin, presented with hypotonia and absent tendon reflexes. The parents described lower limb muscle weakness since the age of 1 month. SMN1 gene analysis confirmed the SMA type 1 diagnosis in this patient carrying no SMN1 copy and 2 SMN2 copies. The approval for gene therapy was rapidly obtained in October 2020 from the French national expert board in an early access program (“Autorisation Temporaire d’Utilisation” for temporary marketing authorization). Gene therapy infusion and clinical and biological monitoring were conducted according to the Protocol for Therapeutic Use.
Results and discussion
After 24 hours of steroid therapy (1 mg/kg per day), an onasemnogene abeparvovec infusion (38.5 mL at 2 × 1013 viral genomes/mL, ie, 1.1 × 1014 vg/kg) was administrated to the patient with SMA at 4 months of age. CHOP-INTEND (Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders) score was 17/64 and HINE (Hammersmith Infant Neurological Examination score) score was 3. Biological results were normal on the day of treatment, including platelet count (562 109/L). Nasopharyngeal multiplex analysis for viruses was negative before the gene therapy infusion. There were no adverse events, and laboratory results were normal at day 3 (D3) after gene therapy. At D8, the patient was lethargic, with vomiting impeding oral steroid administration. Laboratory findings showed thrombocytopenia (<3 109/L), elevated lactate dehydrogenase (LDH) (4836.8 IU/L), and transaminases (aspartate transaminase (AST) level, 329.7 IU/L; alanine aminotransferase (ALT) level, 45.9 IU/L [normal]). From D9 to D12, steroids were well taken orally. Platelet count was then checked on a citrate tube to exclude possible platelet aggregates. At D12, she developed mucocutaneous pallor, oliguria, and edema. Subsequent acute renal failure (serum creatinine level, 228 µmol/L; urea level, 32.8 mmol/L) with hyponatremia (111 mmol/L), hemolytic anemia (hemoglobin level, 4.7 g/dL with schistocytes 6%), thrombocytopenia (22 109/L), and elevated transaminases (AST level, 281.8 IU/L; ALT level, 58.6 IU/L) confirmed the TMA diagnosis (Figure 1B). Hemofiltration was initiated immediately, and steroids were administered by IV infusion. Systemic signs of TMA included pancreatic (elevated lipase level, 384.1 IU/L) and skin damage (Figure 1C). Other investigations demonstrated a negative polymerase chain reaction test for shigatoxin and normal ADAMTS13 activity. At D12, soluble C5b9 complex (sC5b9), a biomarker of complement activation, was increased; the complement levels of C3, C4, and factors H and I (FI) were within normal ranges (Table 1). The sequencing of susceptibility complement genes for atypical hemolytic uremic syndrome (aHUS) identified a variant of unknown significance (VUS), classified benign by PolyPhen in the complement factor I (CFI) gene [c.1322A>G; p.(Lys441Arg)]. The progressive clinical presentation of this patient was suggestive of aHUS. The association between aHUS and AAV gene therapy treatment had not been described in the literature at the time. A decision was made to treat the patient with eculizumab according to the treatment recommendations for pediatric aHUS. After meningococcal vaccination and phenoxymethylpenicilline prophylaxis, she received 3 injections of eculizumab (D13, D19, D40) according to the aHUS treatment protocol for children weighing less than 10 kg (Figure 1).7 Results of monitoring of complement and residual free eculizumab levels are shown in Table 1; CH50 (50% hemolytic complement) was undetectable after eculizumab treatment, and free eculizumab levels targeted the minimum concentration reported in aHUS at D18 (428 µg/L) and D25 (415 µg/L) but not at D40 (81 µg/L) before eculizumab administration. The treatment protocol specifies that plasma concentration of free eculizumab greater than 100 µg/L indicates a marked reduction in CH50 activity. Five days after the first eculizumab injection, C3 was decreased, with normal C4, suggesting a low level of systemic activation of the alternative pathway. The sC5b9 level was normal after the first eculizumab dose. After 1 month, TMA appeared to improve, without renal recovery. We suspect that TMA markers re-increased at D40 (thrombocytopenia, 43 109/L; elevated LDH level, 1542 IU/L; schistocytes 3%; and acute kidney injury with oliguria) because of new-onset Staphylococcus epidermidis sepsis. Staphylococcus epidermidis infection was attributed to prolonged hospitalization, the presence of central catheters required for dialysis, and treatment-induced immunosuppression. Severe dysautonomia compromised the tolerance of hemofiltration. Death occurred after cardiac arrest because of a suspected combination of factors, such as hypovolemia, sepsis, and cardiac dysfunction, in this vulnerable child. No autopsy was performed.
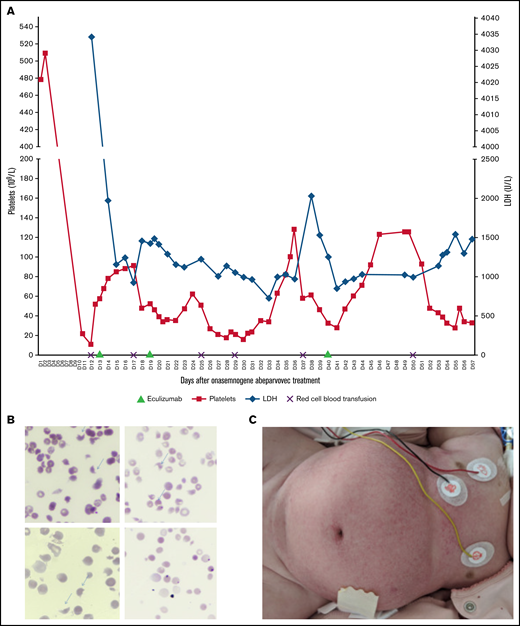
Clinico-biological characteristics of TMA. (A) Trends in platelet and LDH levels during hospitalization. Eculizumab was administrated at days 13, 19, and 40 after onasemnogene abeparvovec infusion. (B) Peripheral smear shows 6% schistocytes (arrows) at D12 (original magnification, ×50). (C) TMA diffuse skin damage.
Clinico-biological characteristics of TMA. (A) Trends in platelet and LDH levels during hospitalization. Eculizumab was administrated at days 13, 19, and 40 after onasemnogene abeparvovec infusion. (B) Peripheral smear shows 6% schistocytes (arrows) at D12 (original magnification, ×50). (C) TMA diffuse skin damage.
Of more than 1400 patients treated with onasemnogene abeparvovec, 9 cases of TMA (including ours) have been reported to date on the US Food and Drug Administration website.6 The EMA (European Medicines Agency) also communicated directly to prescribers about the risk of TMA following onasemnogene abeparvovec.8 This is the first case with a fatal outcome. Three TMA cases induced by this therapy were reported after our current report of this patient.5 Our case shows similarities to the other reported cases: TMA occurred rapidly after treatment (D7-D10) and was predominated by acute renal failure, and further investigations showed an increase in sC5b9. One of the 3 cases previously described showed C4 consumption associated with sC5b9 increase, which may be explained by a classical or lectin pathway. Our patient had increased sC5b9 but no alternate or classical complement pathway consumption. Moreover, 2 of the 3 reported cases had impaired oral steroids consumption caused by vomiting.5 Unlike 2 cases where concomitant infections were diagnosed before or shortly after the infusion, our patient had no TMA predisposing factors at the time of treatment such as infections (including negative shigatoxin), anti–factor H antibodies, ADAMTS-13 activity defect, or known genetic susceptibility to HUS. We therefore suspect that complement overactivation is involved in the pathogenesis of gene therapy–related TMA, as seen in bone marrow transplantation–related TMA. The identified VUS variant in complement factor I has been reported in 1 patient with aHUS and in patients with age-related macular degeneration (AMD) with low and normal FI levels. Rare genetic variants in the CFI gene are associated with aHUS and advanced AMD and commonly result in reduced serum FI levels.9-11 The FI dosing is highly variable in plasma, leading to challenges in the interpretation of the functional consequences of the rare variants identified in the CFI gene.12 In addition, a normal FI level does not exclude a functional deficiency. The variant is located in the serine protease (SP) domain, which allows substrate recognition and cleavage of C3b or C4b in the presence of a cofactor such as factor H. Variants found in the SP domain could interfere with complement regulation. Further studies are needed to draw conclusions on the pathogenicity of this variant. However, our observation suggests that onasemnogene abeparvovec may be the “second hit” in a genetic background that predisposes to the occurrence and the severity of TMA clinical symptoms, as described after hematopoietic stem cell transplant.13
Thrombocytopenia, or TMA with complement activation, has recently been reported in other AAV9-based gene therapies such as in Duchenne muscular dystrophy (4 cases of TMA reported of 15 patients treated in clinical trials14 ) and in Danon disease (1 case of TMA of 2 patients treated at high dose [1.1 × 1014 vg/kg]15 ). In most reported cases, however, the outcome was favorable.
Complement dysregulation leading to C5b9 deposition on endothelial cells is a well-described mechanism of aHUS. The increase in sC5b9 is seen in complement-related diseases such as C3 glomerulonephritis and aHUS. It is also proposed as a biomarker of disease progression in typical HUS or TMA after hematopoietic stem cell transplantation.16,17 Eculizumab, the recommended treatment for complement-associated TMA, had mixed effects on the laboratory results in our patient: a slight increase in platelet count and a decrease in LDH level, without signs of clinical improvement. In 1 of the reported US cases, a child was treated with a single dose of eculizumab, although the efficacy of the treatment was not described.5 Therefore, although eculizumab may be effective in gene therapy–induced TMA, further investigation is warranted.
In summary, TMA induced by AAV gene therapy is a serious, potentially life-threatening complication. Parents must be informed of this risk. Evaluation of predisposing factors for TMA (infections [including negative shigatoxin], anti–factor H antibodies, ADAMTS-13 activity defect or known genetic susceptibility to HUS, sC5b9 dosage) should be undertaken before treatment if possible. Complement testing (C3, C4, CH50, sC5b9, factor H, FI) in pretreatment screening seems important to assess kinetics after treatment. The risk-benefit ratio must be assessed according to patient phenotype and the course of the underlying disease. Early recognition of TMA is absolutely essential for ongoing patient management. We recommend posttreatment follow-up ensuring that steroids are appropriately administered and close serial monitoring of TMA markers to adapt the treatment and limit disease course. The ongoing challenge of AAV gene therapy is to better understand the pathophysiologic mechanisms underlying this immunotoxicity and to find an effective safe targeted immunotherapy that minimizes patient risks.
Acknowledgments
The authors thank the patient’s family who accepted this publication. They also thank Jean-Michel Liet, Pascale Saugier-Veber, Nadezhda Roumeliotis, Guillaume Decompte, and Benjamin Cogné.
Authorship
Contribution: J.G. and A.d.P. collected and interpreted data, wrote the manuscript, and provided figures; F.P., V.F.-B., E.A.-L., C.D., M.D., Y.P., and C.B. collected and interpreted data; I.D. and G.R. collected and interpreted data and supervised the study; and S.M. collected and interpreted data, wrote the manuscript, and supervised the study.
Conflict-of-interest disclosure: V.F.-B. has received fees from Alexion Pharmaceuticals, Roche, and Apellis for invited lectures and/or board membership and is the recipient of a research grant from Alexion Pharmaceuticals. I.D. has received fees from Avexis, Roche, PTC Inc, Sarepta, and Biogen for invited lectures and scientific board meeting. Y.P. has received fees from Avexis, Roche, PTC Therapeutics, Biogen, and Novartis for invited lectures and/or scientific board meetings. The remaining authors declare no competing financial interests.
Correspondence: Sandra Mercier, CHU Nantes, Service de Génétique Médicale, 44000 Nantes, France; e-mail: sandra.mercier@chu-nantes.fr.


