Key Points
C5a/C5aR1 signaling in endothelial cells is a common prothrombogenic effector spanning from rare genetic diseases and viral infections.
C5a causes RalA-mediated exocytosis of vWF and P-selectin, which favor further vWF binding on the endothelium and platelet aggregates.

Abstract
Unrestrained activation of the complement system till the terminal products, C5a and C5b-9, plays a pathogenetic role in acute and chronic inflammatory diseases. In endothelial cells, complement hyperactivation may translate into cell dysfunction, favoring thrombus formation. The aim of this study was to investigate the role of the C5a/C5aR1 axis as opposed to C5b-9 in inducing endothelial dysfunction and loss of antithrombogenic properties. In vitro and ex vivo assays with serum from patients with atypical hemolytic uremic syndrome (aHUS), a prototype rare disease of complement-mediated microvascular thrombosis due to genetically determined alternative pathway dysregulation, and cultured microvascular endothelial cells, demonstrated that the C5a/C5aR1 axis is a key player in endothelial thromboresistance loss. C5a added to normal human serum fully recapitulated the prothrombotic effects of aHUS serum. Mechanistic studies showed that C5a caused RalA-mediated exocytosis of von Willebrand factor (vWF) and P-selectin from Weibel-Palade bodies, which favored further vWF binding on the endothelium and platelet adhesion and aggregation. In patients with severe COVID-19 who suffered from acute activation of complement triggered by severe acute respiratory syndrome coronavirus 2 infection, we found the same C5a-dependent pathogenic mechanisms. These results highlight C5a/C5aR1 as a common prothrombogenic effector spanning from genetic rare diseases to viral infections, and it may have clinical implications. Selective C5a/C5aR1 blockade could have advantages over C5 inhibition because the former preserves the formation of C5b-9, which is critical for controlling bacterial infections that often develop as comorbidities in severely ill patients. The ACCESS trial registered at www.clinicaltrials.gov as #NCT02464891 accounts for the results related to aHUS patients treated with CCX168.
Introduction
The endothelium, which lines the vessels at the interface with blood flow, is crucial in maintaining vascular homeostasis. Under physiological conditions, the luminal surface of endothelial cells possesses anticoagulant and antithrombogenic properties that inhibit platelet adhesion and prevent the activation of the coagulation system.1 Activated or injured endothelial cells lose their antithrombogenic properties, thus leading to thrombus formation.
Endothelial dysfunction has been described in several diseases, including diabetes, atherosclerosis, and infections, which are major causes of morbidity and mortality.2 Endothelial cells are the target of inflammatory and immune mediators, including the activation products of the complement cascade.
The complement system, a component of innate immunity that is organized into three activation pathways (classical, lectin, and alternative pathways), is the first response to invading pathogens.3,4 Activation of the complement system ends with the formation of terminal products: the anaphylatoxin C5a, which attracts and activates neutrophils and macrophages, and the membrane attack complex C5b-9, which forms a lysis-promoting pore in bacteria.4,5 To avoid unwanted damage to self-tissues, complement activation is finely regulated. Unrestrained activation of the complement system plays a pathogenetic role in acute and chronic inflammatory diseases.3 At the endothelial cell level, complement dysregulation may translate into cell dysfunction, thrombus formation, and intravascular coagulation.
In this context, several studies on atypical hemolytic uremic syndrome (aHUS), a rare disease characterized by the extensive formation of thrombi in the microcirculation of the kidney and other organs due to genetically determined dysregulation of the complement alternative pathway,6 have contributed to highlighting the role of complement activation products in microvascular thrombosis. By using an ex vivo test in which microvascular endothelial cells were incubated with serum from patients with aHUS, we documented that aHUS serum, but not ctr serum, induced intense C5b-9 endothelial deposits.7,8 Similarly, with another ex vivo assay based on TF1 PIGAnull cells, an engineered cell line that is devoid of the cell-surface complement regulators CD55 and CD59, other authors documented excessive cellular deposition of C5b-9 after exposure to aHUS serum.9
The role of C5 activation products in aHUS has been confirmed by clinical success of the anti-C5 monoclonal antibody eculizumab, which radically improved the prognosis of aHUS by inducing and maintaining disease remission.10,11 C5 inhibition has been shown to be effective in reducing thrombotic risk in other diseases characterized by defective control of complement activation, including paroxysmal nocturnal hemoglobinuria.11,12 Recently, eculizumab has been proposed as a therapeutic tool for limiting the exaggerated complement activation that is associated with widespread endothelial damage and systemic hypercoagulopathy that characterize severe forms of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) respiratory infections (COVID-19),13 although results are conflicting, and a trial with another anti-C5 antibody, ravulizumab, was halted by the company (https://ir.alexion.com/news-releases/news-release-details/alexion-provides-update-phase-3-study-ultomirisr-ravulizumab).
However, treatment with C5-blocking agents has several disadvantages. The drug must be administered intravenously chronically. In addition, blocking C5 activation prevents the formation of both terminal pathway products, C5a and C5b-9, thus exposing patients to the risk of developing infections. Studies in a mouse model of aHUS showed that C5aR1 and C5b-9 pathways mediated different aspects of the disease.14 A better understanding of the role of C5a/C5aR1 and C5b-9 in microvascular thrombosis in humans could be instrumental in defining therapeutic approaches that will specifically target either of the two terminal complement products, thus limiting the risk of infections.
The aim of this study was to investigate the role of the C5a/C5aR1 axis as opposed to C5b-9 in inducing endothelial dysfunction and loss of antithrombogenic properties.
Through in vitro and ex vivo assays with serum from patients with aHUS used as a prototype disease of complement-mediated microvascular thrombosis, we demonstrated the central role that C5a/C5aR1 interaction plays in loss of endothelial thromboresistance in humans. We also found that C5a combined with normal human serum recapitulated the prothrombotic effects of aHUS serum on endothelial cells.
Through mechanistic studies, we documented that C5a directly induces exocytosis of von Willebrand factor (vWF) and P-selectin from Weibel-Palade bodies (WPBs) of endothelial cells, which favor further vWF binding on the endothelium. In COVID-19, we found the same C5a-dependent pathogenic mechanism, which supports the hypothesis that the C5a/C5aR1 axis is a common prothrombogenic effector spanning from genetic rare diseases to viral infections.
Methods
Study subjects
In this study, we analyzed serum samples from 11 patients with aHUS. Serum samples were collected during full or hematological (normalization of hematological parameters with renal dysfunction) remission. The patients’ characteristics are summarized in Table 1. We enrolled patients who participated in the study entitled “Genetic and biochemical abnormalities in hemolytic uremic syndrome and thrombotic thrombocytopenic purpura,” which aimed to identify the genetic and biochemical factors that predispose to the development (and relapse) of aHUS, thrombotic thrombocytopenia purpura (TTP), and severe typical HUS in patients referred to the International Registry of HUS/TTP.
Five of the patients with aHUS in hematological remission (patients 4, 7, 8, 10, and 11 in Table 1) were also enrolled in the ACCESS study. ACCESS was an open-label phase 2 study that aimed to assess the effect of C5aR1 antagonist therapy through CCX168 on ex vivo thrombus formation in patients with end stage renal disease and aHUS. Treatment (30 mg CCX168 by oral administration, twice daily for 2 weeks) was started the day after baseline evaluations. We analyzed serum samples collected at the start of the first dialysis session (baseline evaluation, time 0) and at the start of dialysis on day 14 (final treatment evaluation).
The above studies were conducted at Clinical Research Center for Rare Diseases Aldo e Cele Daccò of the Istituto di Ricerche Farmacologiche Mario Negri IRCCS in Ranica (Bergamo), Italy.
We also studied 6 patients with COVID-19 who were admitted to the COVID Unit of the Azienda Socio Sanitaria Territoriale (ASST) Papa Giovanni XXIII Hospital in Bergamo, Italy, between March and April 2020 because of severe respiratory distress due to COVID-19 and who were receiving continuous positive airway pressure (CPAP) ventilator support from 24 hours to 5 days. The diagnosis of COVID-19 was based on the 19 March 2020 WHO interim guidance criteria.15 The diagnosis was confirmed by detection, at admission, of the SARS-CoV-2 genome on nasal swabs and respiratory samples. In patients with COVID-19, no clinical signs of thrombotic microangiopathies and no pathogenic variants in complement genes have been found. Patients' characteristics are summarized in Table 2.
Twenty healthy subjects were also recruited as controls. Sera from 10 healthy subjects were pooled and used as control serum (ctr). Ten healthy subjects were used as blood donors for thrombus formation experiments.
All studies were approved by the Ethical Committee of the Azienda Sanitaria Locale Bergamo, Italy. Written informed consent was obtained from enrolled patients.
Study assessments
Methods used for evaluating complement deposition, formation of platelet aggregates, and staining of vWF, P-selectin, C5aR1, and CD41 on HMEC-1 as well as serum and plasma C5a levels, scanning electron microscopy, and statistical analysis are detailed in supplemental Methods.
Results
Serum from patients with aHUS induces platelet adhesion and aggregation on microvascular endothelial cells through C5a/C5aR1
Incubation of adenosine 5′-diphosphate (ADP)-activated HMEC-1 with serum from patients with aHUS (Table 1) collected in remission out of treatment induced 3- to 4-fold elevated C3 and C5b-9 deposits, compared with serum from healthy subjects (ctr serum pool), confirming our previous results (supplemental Figures 1A-C and 2A-D).7,8
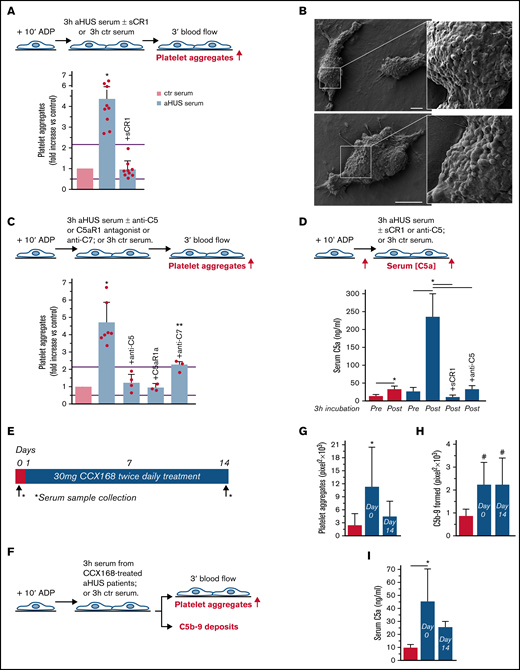
To evaluate whether complement activation resulted in loss of endothelial thromboresistance, HMEC-1 were incubated with aHUS serum or ctr serum and then perfused with heparinized whole blood (Figure 1A). HMEC-1 preexposed to aHUS serum had a 5-fold wider cell surface area covered by platelet aggregates compared with cells exposed to ctr serum (Figure 1A; representative images in supplemental Figure 3). Notably, all fold increase values obtained with aHUS serum were higher than the upper limit of normal range (calculated as mean ± 2 standard deviation (SD) of results obtained with sera from 10 single healthy subjects; supplemental Figure 4). Adding sCR1 (Figure 1A) or an anti-C5 antibody (Figure 1C) to aHUS serum reduced the area covered by platelet aggregates to values observed with ctr serum pool, indicating that the terminal pathway plays a key role. Scanning electron microscopy evaluation of HMEC-1 exposed to aHUS serum and then perfused with heparinized whole blood documented the attachment of platelets to the endothelial cell monolayer to form organized aggregates (Figure 1B; supplemental Figure 5). As shown in supplemental Figure 6, immunofluorescence staining with anti-CD41 and anti-vWF confirmed that aggregates mainly consisted of platelets and vWF.

Serum from patients with aHUS induces platelet adhesion and aggregation on microvascular endothelial cells through C5a/C5aR1 signaling. (A, upper panel) Experimental design. HMEC-1 were activated with ADP, exposed for 3 hours to ctr serum or serum from patients with aHUS in remission, and then perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. aHUS serum was added or not with the pan complement inhibitor sCR1 (150 μg/mL). Formation of platelet aggregates was evaluated at the end of blood perfusion (through confocal microscopy). (A, lower panel) Endothelial surface area with positive staining for platelet aggregates. Results are shown as fold increase of stained surface area after incubation with ctr serum run in parallel. Data are mean ± SD; n = 9 independent experiments. Red points represent fold increase values of single experiments. Purple lines are the upper and lower limits of normal range. (B) Representative images of the ultrastructure of aggregates of platelets adhered on HMEC-1 preexposed to aHUS serum, evaluated with scanning electron microscopy at low magnification (left panels), with the corresponding high magnification insets (right panels). Insets show high-power view of same platelet aggregate. Scale bars represent 10 µm (n = 2 experiments). (C, upper panel) Experimental design. HMEC-1 were activated with ADP, exposed for 3 hours to ctr or aHUS serum, and then perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. aHUS serum was added or not, with an anti-C5 antibody (135 μg/mL), or the C5aR1 antagonist CCX168 (200 ng/mL), or an anti-C7 antibody (350 μg/mL). Formation of platelet aggregates was evaluated at the end of the blood perfusion. (C, lower panel) Endothelial surface area covered by platelet aggregates. Results are shown as fold increase of stained surface area after incubation with ctr serum run in parallel. Data are mean ± SD; n = 4 independent experiments for +anti-C5; n = 3 independent experiments for +C5aR1a and +anti-C7. Red points represent fold increase values of single experiments. Purple lines are the upper and lower limits of normal range. (D, upper panel) Experimental design. HMEC-1 were activated with ADP and then exposed for 3 hours to ctr or aHUS serum. aHUS serum was added or not with sCR1 (150 μg/mL) or the anti-C5 antibody (135 μg/mL). Serum C5a levels were measured before and after incubation. (D, lower panel) C5a concentration in ctr or aHUS serum before and after incubation with ADP-activated HMEC-1. Data are mean ± SD; n = 3 independent experiments. (E) Scheme of treatment with the C5a receptor antagonist CCX168 and serum sample collection in patients with aHUS enrolled in the ACCESS study. (F) Experimental design. HMEC-1 were activated with ADP, exposed for 3 hours to ctr serum or serum from patients with aHUS (taken before treatment with CCX168, day 0, and after 14 days of treatment), and then analyzed for C5b-9 deposits or perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. Formation of platelet aggregates was evaluated at the end of blood perfusion. (G-H) Endothelial surface area with positive staining for platelet aggregates (G) or C5b-9 (H) after incubation of ADP-activated HMEC-1 with ctr serum or serum from patients with aHUS treated with CCX168 collected at days 0 and 14 of the study. Data are mean ± SD of cumulative data from 5 patients. (I) C5a levels in ctr serum or in serum from patients with aHUS taken before treatment with CCX168, day 0, and after 14 days of treatment (n = 5). *P < .05 vs the groups indicated by horizontal bar or, if not indicated, vs all groups. **P < .05 vs +C5aR1a. #P < .05 vs ctr serum.
Serum from patients with aHUS induces platelet adhesion and aggregation on microvascular endothelial cells through C5a/C5aR1 signaling. (A, upper panel) Experimental design. HMEC-1 were activated with ADP, exposed for 3 hours to ctr serum or serum from patients with aHUS in remission, and then perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. aHUS serum was added or not with the pan complement inhibitor sCR1 (150 μg/mL). Formation of platelet aggregates was evaluated at the end of blood perfusion (through confocal microscopy). (A, lower panel) Endothelial surface area with positive staining for platelet aggregates. Results are shown as fold increase of stained surface area after incubation with ctr serum run in parallel. Data are mean ± SD; n = 9 independent experiments. Red points represent fold increase values of single experiments. Purple lines are the upper and lower limits of normal range. (B) Representative images of the ultrastructure of aggregates of platelets adhered on HMEC-1 preexposed to aHUS serum, evaluated with scanning electron microscopy at low magnification (left panels), with the corresponding high magnification insets (right panels). Insets show high-power view of same platelet aggregate. Scale bars represent 10 µm (n = 2 experiments). (C, upper panel) Experimental design. HMEC-1 were activated with ADP, exposed for 3 hours to ctr or aHUS serum, and then perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. aHUS serum was added or not, with an anti-C5 antibody (135 μg/mL), or the C5aR1 antagonist CCX168 (200 ng/mL), or an anti-C7 antibody (350 μg/mL). Formation of platelet aggregates was evaluated at the end of the blood perfusion. (C, lower panel) Endothelial surface area covered by platelet aggregates. Results are shown as fold increase of stained surface area after incubation with ctr serum run in parallel. Data are mean ± SD; n = 4 independent experiments for +anti-C5; n = 3 independent experiments for +C5aR1a and +anti-C7. Red points represent fold increase values of single experiments. Purple lines are the upper and lower limits of normal range. (D, upper panel) Experimental design. HMEC-1 were activated with ADP and then exposed for 3 hours to ctr or aHUS serum. aHUS serum was added or not with sCR1 (150 μg/mL) or the anti-C5 antibody (135 μg/mL). Serum C5a levels were measured before and after incubation. (D, lower panel) C5a concentration in ctr or aHUS serum before and after incubation with ADP-activated HMEC-1. Data are mean ± SD; n = 3 independent experiments. (E) Scheme of treatment with the C5a receptor antagonist CCX168 and serum sample collection in patients with aHUS enrolled in the ACCESS study. (F) Experimental design. HMEC-1 were activated with ADP, exposed for 3 hours to ctr serum or serum from patients with aHUS (taken before treatment with CCX168, day 0, and after 14 days of treatment), and then analyzed for C5b-9 deposits or perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. Formation of platelet aggregates was evaluated at the end of blood perfusion. (G-H) Endothelial surface area with positive staining for platelet aggregates (G) or C5b-9 (H) after incubation of ADP-activated HMEC-1 with ctr serum or serum from patients with aHUS treated with CCX168 collected at days 0 and 14 of the study. Data are mean ± SD of cumulative data from 5 patients. (I) C5a levels in ctr serum or in serum from patients with aHUS taken before treatment with CCX168, day 0, and after 14 days of treatment (n = 5). *P < .05 vs the groups indicated by horizontal bar or, if not indicated, vs all groups. **P < .05 vs +C5aR1a. #P < .05 vs ctr serum.
To identify which of the two terminal activation products, C5a or C5b-9, was mainly involved in platelet adhesion and aggregation, the above experiments were repeated in the presence of a specific C5aR1 antagonist, CCX168 (Avacopan, Chemocentrix), or an anti-C7 antibody (Figure 1C, upper panel) that we previously reported to be effective in blocking aHUS serum-induced C5b-9 deposits on HMEC-1.7 CCX168 was used at the concentration of 200 ng/mL, corresponding to plasmatic Cmax levels reported in patients with antineutrophil cytoplasmic antibody vasculitis.16 CCX168 fully normalized the area covered by platelet aggregates on cells exposed to aHUS serum, whereas the anti-C7 antibody exerted only a partial inhibitory effect (Figure 1C, lower panel; representative images in supplemental Figure 3). In further experiments, we tested the effect of different concentrations of CCX168 (from 30-600 ng/mL; supplemental Figure 7A) added to aHUS serum. As shown in supplemental Figure 7B, a significant reduction of area covered by platelet aggregates was already observed with 80 ng/mL CCX168, corresponding to trough levels reported in the published trial.16 C5b-9 deposition induced by aHUS serum was not inhibited by CCX168 (supplemental Figure 7C).
We observed an approximately 10-fold increase in C5a levels in aHUS serum at the end of incubation with HMEC-1, compared with preincubation levels, whereas the concentration of C5a only doubled in ctr serum (Figure 1D), confirming that C5a is generated in high amounts during the incubation of aHUS serum with HMEC-1. C5a could derive from both activation of C5 in serum and activation of C5 released from endothelial cells.17
We then moved to test the effect of in vivo inhibition of the C5a/C5aR1 axis on ex vivo formation of platelet aggregates by analyzing the prothrombogenic effect of serum from patients with aHUS, all in stable hematological remission, who received 2-week treatment with CCX168 (Figure 1E-F). Exposure of ADP-activated HMEC-1 to serum collected before treatment resulted in massive platelet adhesion and aggregation when the cells were perfused with ctr whole blood (Figure 1G). When endothelial cells were incubated with serum collected at the end of CCX168 treatment, ex vivo formation of platelet aggregates normalized, confirming that C5a/C5aR1 plays a key role in the aHUS serum prothrombotic effect (Figure 1G). In vivo treatment with CCX168 had no effect on C5b-9 formed on HMEC-1 exposed to aHUS serum (Figure 1H). C5a levels were higher in serum from patients with aHUS than in serum from healthy subjects and showed a trend to decrease (albeit not significantly) at the end of the 2-week treatment with CCX168 (Figure 1I).
Excess C5a added to normal serum induces platelet adhesion and aggregation on microvascular endothelial cells
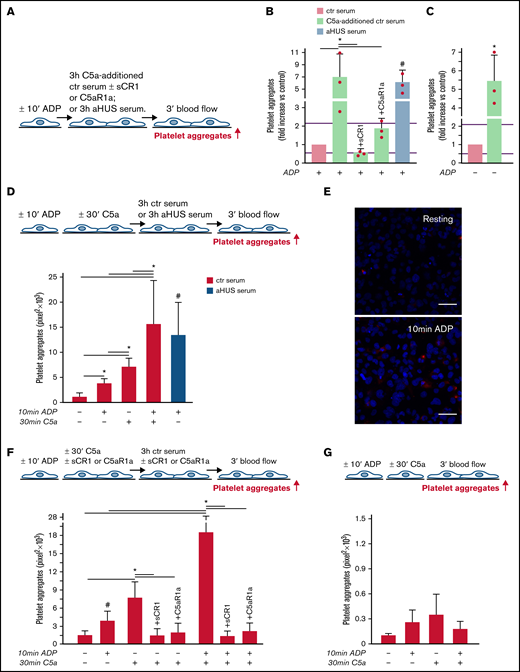
aHUS is associated with genetically determined dysregulation of the alternative pathway.6 To assess whether C5a may exert a prothrombotic effect on endothelial cells in conditions of normal regulation of the alternative pathway, thrombus formation experiments were repeated with C5a added to normal human serum (Figure 2A). The concentration of C5a added (200 ng/mL) was in the range of the C5a levels measured in the aHUS serum after incubation on ADP-activated endothelial cells (Figure 1D). C5a-added ctr serum induced massive platelet aggregation on ADP-activated HMEC-1 so that the cell surface area covered by platelet aggregates was ∼7-fold wider than with ctr serum alone and comparable to that induced by aHUS serum (Figure 2B). Platelet adhesion and aggregation induced by C5a-added serum on HMEC-1 was significantly reduced by sCR1 or by the C5aR1 antagonist CCX168 (Figure 2B). C5a-added ctr serum also induced a significant increase in formation of platelet aggregates on HMEC-1 that had not been preexposed to ADP (resting HMEC-1) compared with ctr serum alone (Figure 2C).

C5a and normal human serum (ctr) induces platelet adhesion and aggregation on HMEC-1. (A) Experimental design. HMEC-1, ADP-activated or resting, were exposed for 3 hours to ctr serum with or without the addition of C5a (200 ng/mL) or to aHUS serum, and then perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. C5a-additioned serum was tested in the presence or absence of sCR1 (150 μg/mL) or the C5aR1 antagonist CCX168 (200 ng/mL). Formation of platelet aggregates was evaluated at the end of the blood perfusion. (B-C) Effect of C5a-additioned ctr serum on formation of platelet aggregates on HMEC-1 (B, ADP-activated; C, resting). Results are shown as fold increase of stained surface area after incubation with ctr serum alone run in parallel. Data are mean ± SD; n = 3 independent experiments. Red points represent fold increase values of single experiments. Purple lines are the upper and lower limits of normal range. *P < .05 vs the groups indicated by horizontal bar. #P < .05 vs ctr serum. (D, upper panel) Experimental design. HMEC-1 were activated or not with ADP for 10 minutes and then stimulated or not with C5a for 30 minutes, exposed for 3 hours to ctr serum, and perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. On selected slides, ADP-activated cells were exposed to aHUS serum, taken as the positive control. Formation of platelet aggregates was evaluated at the end of the blood perfusion. (D, lower panel) Endothelial surface area covered by platelet aggregates after incubation with ctr serum of resting (no ADP, no C5a), ADP-activated, C5a-stimulated or ADP-activated/C5a-stimulated HMEC-1; or after incubation with aHUS serum. Data are mean ± SD; n = 4 independent experiments. *P < .05 vs the groups indicated by horizontal bar. #P < .05 vs resting and ADP-activated HMEC-1 + ctr serum. (E) Representative images of immunostaining for C5aR1 (red staining) on resting or ADP-activated HMEC-1. Blue: DAPI staining. Original magnification: ×400. Scale bar represents 50 μm. (F, upper panel) Experimental design. HMEC-1 were activated or not with ADP for 10 minutes and then stimulated or not with C5a for 30 minutes, exposed for 3 hours to ctr serum, and perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. In 3 of 4 experiments, additional samples were run in which either sCR1 (150 μg/mL) or the C5aR1 antagonist CCX168 (200 ng/mL) were added during the C5a stimulation and the exposure to serum. Formation of platelet aggregates was evaluated at the end of blood perfusion. (F, lower panel) Endothelial surface area covered by platelet aggregates after incubation with ctr serum of resting, ADP-activated, C5a-stimulated or ADP-activated/C5a-stimulated HMEC-1. Data are mean ± SD; n = 3 to 4 independent experiments. *P < .05 vs the groups indicated by horizontal bar. #P < .05 vs resting. (G, upper panel) Experimental design. HMEC-1 were activated or not with ADP for 10 minutes and then stimulated or not with C5a for 30 minutes and perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. Formation of platelet aggregates was evaluated at the end of blood perfusion. (G, lower panel) Endothelial surface area covered by platelet aggregates on resting, ADP-activated, C5a-stimulated or ADP-activated/C5a-stimulated HMEC-1. Data are mean ± SD; n = 3 independent experiments.
C5a and normal human serum (ctr) induces platelet adhesion and aggregation on HMEC-1. (A) Experimental design. HMEC-1, ADP-activated or resting, were exposed for 3 hours to ctr serum with or without the addition of C5a (200 ng/mL) or to aHUS serum, and then perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. C5a-additioned serum was tested in the presence or absence of sCR1 (150 μg/mL) or the C5aR1 antagonist CCX168 (200 ng/mL). Formation of platelet aggregates was evaluated at the end of the blood perfusion. (B-C) Effect of C5a-additioned ctr serum on formation of platelet aggregates on HMEC-1 (B, ADP-activated; C, resting). Results are shown as fold increase of stained surface area after incubation with ctr serum alone run in parallel. Data are mean ± SD; n = 3 independent experiments. Red points represent fold increase values of single experiments. Purple lines are the upper and lower limits of normal range. *P < .05 vs the groups indicated by horizontal bar. #P < .05 vs ctr serum. (D, upper panel) Experimental design. HMEC-1 were activated or not with ADP for 10 minutes and then stimulated or not with C5a for 30 minutes, exposed for 3 hours to ctr serum, and perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. On selected slides, ADP-activated cells were exposed to aHUS serum, taken as the positive control. Formation of platelet aggregates was evaluated at the end of the blood perfusion. (D, lower panel) Endothelial surface area covered by platelet aggregates after incubation with ctr serum of resting (no ADP, no C5a), ADP-activated, C5a-stimulated or ADP-activated/C5a-stimulated HMEC-1; or after incubation with aHUS serum. Data are mean ± SD; n = 4 independent experiments. *P < .05 vs the groups indicated by horizontal bar. #P < .05 vs resting and ADP-activated HMEC-1 + ctr serum. (E) Representative images of immunostaining for C5aR1 (red staining) on resting or ADP-activated HMEC-1. Blue: DAPI staining. Original magnification: ×400. Scale bar represents 50 μm. (F, upper panel) Experimental design. HMEC-1 were activated or not with ADP for 10 minutes and then stimulated or not with C5a for 30 minutes, exposed for 3 hours to ctr serum, and perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. In 3 of 4 experiments, additional samples were run in which either sCR1 (150 μg/mL) or the C5aR1 antagonist CCX168 (200 ng/mL) were added during the C5a stimulation and the exposure to serum. Formation of platelet aggregates was evaluated at the end of blood perfusion. (F, lower panel) Endothelial surface area covered by platelet aggregates after incubation with ctr serum of resting, ADP-activated, C5a-stimulated or ADP-activated/C5a-stimulated HMEC-1. Data are mean ± SD; n = 3 to 4 independent experiments. *P < .05 vs the groups indicated by horizontal bar. #P < .05 vs resting. (G, upper panel) Experimental design. HMEC-1 were activated or not with ADP for 10 minutes and then stimulated or not with C5a for 30 minutes and perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. Formation of platelet aggregates was evaluated at the end of blood perfusion. (G, lower panel) Endothelial surface area covered by platelet aggregates on resting, ADP-activated, C5a-stimulated or ADP-activated/C5a-stimulated HMEC-1. Data are mean ± SD; n = 3 independent experiments.
In additional experiments, resting or ADP-activated HMEC-1 were stimulated for 30 minutes with C5a, thereafter exposed for 3 hours to ctr serum, and then perfused with whole blood (Figure 2D). Resting HMEC-1 stimulated with C5a, followed by ctr serum, exhibited a greater surface area covered by platelet aggregates than resting and ADP-activated HMEC-1 exposed to ctr serum (Figure 2D). ADP-activated HMEC-1 stimulated with C5a followed by ctr serum showed massive platelet adhesion and formation of platelet aggregates, which was significantly higher than that observed with either ADP or C5a, suggesting a synergistic effect (Figure 2D). In this context, it is important to note that 10-minute ADP-activated HMEC-1 upregulated C5aR1 expression (Figure 2E). sCR1 and CCX168 fully prevented formation of platelet aggregates on C5a-stimulated and ADP-activated/C5a-stimulated HMEC-1 (Figure 2F; representative images in supplemental Figure 8).
The above experiments were repeated without the step with ctr serum. The area covered by platelet aggregates was negligible on HMEC-1 stimulated with C5a, even when C5a stimulation was combined with ADP preactivation (Figure 2G).
Overall, the above results indicate that the stimulation of C5a/C5aR1 signaling exerts a prothrombotic effect on microvascular endothelial cells, and the presence of serum molecules is necessary for C5a/C5aR1 to induce massive formation of platelet aggregates.
Role of vWF and P-selectin in C5a-induced platelet adhesion and aggregation on microvascular endothelial cells
We then investigated the mechanism(s) underlying the prothrombogenic action of C5a/C5aR1. We focused on WPBs, based on our earlier studies documenting that exposure to C5a stimulated vWF release from HMEC-1.18
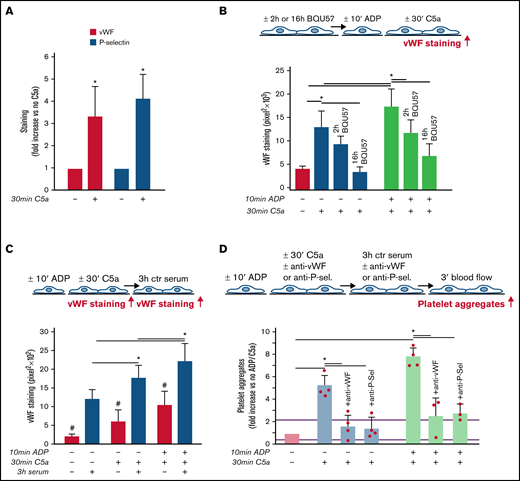
As shown in Figure 3A, C5a stimulation led to a significant increase in vWF staining on endothelial cells, which was fully suppressed by coincubation with CCX168 (supplemental Figure 9). On C5a-stimulated HMEC-1, P-selectin staining was also higher than on resting cells (Figure 3A), which indicated that C5a induced the release of WPBs. To confirm that C5a stimulated WPB exocytosis, vWF staining was repeated on C5a-stimulated HMEC-1, which were preincubated with BQU57, a specific inhibitor of RalA, a small GTPase central to the molecular machinery involved in WPB exocytosis (Figure 3B).19-21 BQU57 significantly reduced vWF staining on C5a-stimulated and on ADP-activated/C5a-stimulated HMEC-1.

Role of vWF and P-selectin in C5a-induced platelet adhesion and aggregation on HMEC-1. (A) Endothelial surface area stained for vWF or P-selectin on resting or C5a-stimulated HMEC-1. Results are shown as fold increase of stained area on resting HMEC-1. Data are mean ± SD; n = 4 (vWF) or 3 (P-selectin) independent experiments. *P < .05 vs no stimulation. (B, upper panel) Experimental design. Before the experiment, HMEC-1 were left for 2 hours or 16 hours with medium added or not with the RalA inhibitor BQU57 (10 µM). Thereafter, HMEC-1 were activated or not for 10 minutes with ADP and then stimulated or not for 30 minutes with C5a (200 ng/mL). vWF staining was performed at the end of C5a stimulation. (B, lower panel) Endothelial surface area stained for vWF. Data are mean ± SD of 13 fields; n = 1 to 2 experiments. *P < .05 vs the groups indicated by horizontal bar. (C, upper panel) Experimental design. HMEC-1 were activated or not with ADP and then stimulated or not with C5a and exposed or not for 3 hours to ctr serum. vWF staining was performed at the end of C5a stimulation or exposure to serum. (C, lower panel) Endothelial surface area stained for vWF. Mean ± SD of 13 fields. *P < .05. #P < .05 vs the corresponding condition with serum. (D, upper panel) Experimental design. HMEC-1 were activated or not with ADP and then stimulated or not with C5a, exposed for 3 hours to ctr serum and then perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. In 3 of 4 experiments, either an anti-vWF (10 μg/mL) or an anti-P-selectin (20 μg/mL) antibody was added during the C5a stimulation and the exposure to serum. Formation of platelet aggregates was evaluated at the end of blood perfusion. (D, lower panel) Endothelial surface area covered by platelet aggregates on resting (no ADP, no C5a), C5a-stimulated, or ADP-activated/C5a-stimulated HMEC-1 incubated with ctr serum. Results are shown as fold increase of stained surface area on resting HMEC-1 incubated with ctr serum alone run in parallel. Data are mean ± SD; n = 3 to 4 independent experiments. Red points represent fold increase values of single experiments. Purple lines are the upper and lower limits of normal range. *P < .05 vs the groups indicated by horizontal bar.
Role of vWF and P-selectin in C5a-induced platelet adhesion and aggregation on HMEC-1. (A) Endothelial surface area stained for vWF or P-selectin on resting or C5a-stimulated HMEC-1. Results are shown as fold increase of stained area on resting HMEC-1. Data are mean ± SD; n = 4 (vWF) or 3 (P-selectin) independent experiments. *P < .05 vs no stimulation. (B, upper panel) Experimental design. Before the experiment, HMEC-1 were left for 2 hours or 16 hours with medium added or not with the RalA inhibitor BQU57 (10 µM). Thereafter, HMEC-1 were activated or not for 10 minutes with ADP and then stimulated or not for 30 minutes with C5a (200 ng/mL). vWF staining was performed at the end of C5a stimulation. (B, lower panel) Endothelial surface area stained for vWF. Data are mean ± SD of 13 fields; n = 1 to 2 experiments. *P < .05 vs the groups indicated by horizontal bar. (C, upper panel) Experimental design. HMEC-1 were activated or not with ADP and then stimulated or not with C5a and exposed or not for 3 hours to ctr serum. vWF staining was performed at the end of C5a stimulation or exposure to serum. (C, lower panel) Endothelial surface area stained for vWF. Mean ± SD of 13 fields. *P < .05. #P < .05 vs the corresponding condition with serum. (D, upper panel) Experimental design. HMEC-1 were activated or not with ADP and then stimulated or not with C5a, exposed for 3 hours to ctr serum and then perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. In 3 of 4 experiments, either an anti-vWF (10 μg/mL) or an anti-P-selectin (20 μg/mL) antibody was added during the C5a stimulation and the exposure to serum. Formation of platelet aggregates was evaluated at the end of blood perfusion. (D, lower panel) Endothelial surface area covered by platelet aggregates on resting (no ADP, no C5a), C5a-stimulated, or ADP-activated/C5a-stimulated HMEC-1 incubated with ctr serum. Results are shown as fold increase of stained surface area on resting HMEC-1 incubated with ctr serum alone run in parallel. Data are mean ± SD; n = 3 to 4 independent experiments. Red points represent fold increase values of single experiments. Purple lines are the upper and lower limits of normal range. *P < .05 vs the groups indicated by horizontal bar.
Additional experiments were performed to evaluate whether the stimulation with C5a favored further binding of circulating vWF on HMEC-1 (Figure 3C). Incubation with ctr serum increased vWF staining on resting HMEC-1. vWF staining after exposure to ctr serum further increased on cells stimulated with C5a alone or in combination with ADP preactivation (Figure 3C).
To assess whether vWF and P-selectin play a role in the prothrombogenic action of C5a, formation of platelet aggregates was evaluated in the presence of anti-vWF or anti-P-selectin antibodies (Figure 3D). Both the anti-vWF and the anti-P-selectin antibodies (added to the cells and to the serum) significantly reduced platelet adhesion and aggregation on HMEC-1 stimulated by C5a or by ADP/C5a followed by exposure to ctr serum.
Role of C5a/C5aR1-induced exocytosis of WPBs in aHUS
We then evaluated whether the above mechanism plays a role in the prothrombogenic effect of aHUS serum. First, we assessed whether the aHUS serum stimulated HMEC-1 to release vWF. Endothelial vWF staining was significantly higher on cells exposed to aHUS serum versus HMEC-1 exposed to ctr serum (Figure 4A). Adding CCX168 to aHUS serum completely prevented the increase of vWF staining, indicating that C5a/C5aR1 signaling plays a central role in vWF accumulation induced by aHUS serum on HMEC-1. In addition, an anti-vWF antibody fully prevented the increase in formation of platelet aggregates on HMEC-1 induced by aHUS serum (Figure 4B, absolute values in pixel2 ; representative images in supplemental Figure 10).
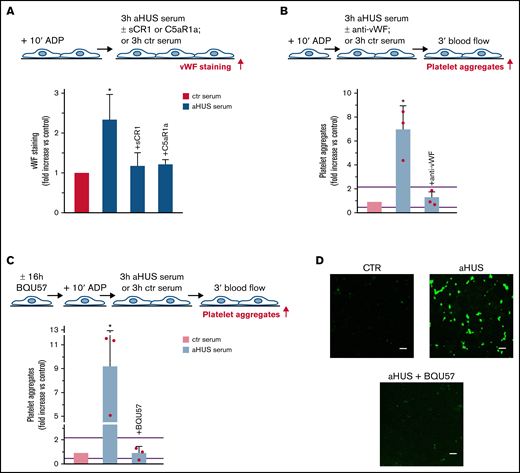
Role of vWF and exocytosis of WPBs in aHUS serum-induced platelet adhesion and aggregation on HMEC-1. (A, upper panel) Experimental design. HMEC-1 were activated with ADP and then exposed for 3 hours to ctr or aHUS serum. aHUS serum was added or not with sCR1 (150 μg/mL) or the C5aR1 antagonist CCX168 (200 ng/mL). vWF staining was performed at the end of exposure to serum. (A, lower panel) Endothelial surface area stained for vWF. Results are shown as fold increase of stained area after incubation, with aHUS serum (n = 4) vs ctr serum run in parallel. Data are mean ± SD. *P < .05 vs all groups. (B, upper panel) Experimental design. ADP-activated HMEC-1 were exposed for 3 hours to ctr or aHUS serum and then perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. aHUS serum was added or not with an anti-vWF antibody (10 μg/mL). Formation of platelet aggregates was evaluated at the end of blood perfusion. (B, lower panel) Endothelial surface area covered by platelet aggregates. Results are shown as fold increase of surface area covered by platelet aggregates after incubation with aHUS serum (n = 3) vs ctr serum run in parallel. Data are mean ± SD. Red points represent fold increase values of single patients. Purple lines are the upper and lower limits of normal range. *P < .05 vs all groups. (C, upper panel) Experimental design. Before the experiment, HMEC-1 were left for 16 hours in medium with or without the RalA inhibitor BQU57 (10 µM). Thereafter, HMEC-1 were activated for 10 minutes with ADP, exposed for 3 hours to ctr or aHUS serum, and then perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. Formation of platelet aggregates was evaluated at the end of blood perfusion. (C, lower panel) Endothelial surface area covered by platelet aggregates. Results are shown as fold increase of stained surface area after incubation with aHUS serum (n = 3) vs ctr serum run in parallel. Data are mean ± SD. Red points represent fold increase values of single patients. Purple lines are the upper and lower limits of normal range. *P < .05 vs all groups. (D) Representative images of experiments (green staining) relative to Figure 4C. Original magnification ×200. Scale bar represents 50 μm.
Role of vWF and exocytosis of WPBs in aHUS serum-induced platelet adhesion and aggregation on HMEC-1. (A, upper panel) Experimental design. HMEC-1 were activated with ADP and then exposed for 3 hours to ctr or aHUS serum. aHUS serum was added or not with sCR1 (150 μg/mL) or the C5aR1 antagonist CCX168 (200 ng/mL). vWF staining was performed at the end of exposure to serum. (A, lower panel) Endothelial surface area stained for vWF. Results are shown as fold increase of stained area after incubation, with aHUS serum (n = 4) vs ctr serum run in parallel. Data are mean ± SD. *P < .05 vs all groups. (B, upper panel) Experimental design. ADP-activated HMEC-1 were exposed for 3 hours to ctr or aHUS serum and then perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. aHUS serum was added or not with an anti-vWF antibody (10 μg/mL). Formation of platelet aggregates was evaluated at the end of blood perfusion. (B, lower panel) Endothelial surface area covered by platelet aggregates. Results are shown as fold increase of surface area covered by platelet aggregates after incubation with aHUS serum (n = 3) vs ctr serum run in parallel. Data are mean ± SD. Red points represent fold increase values of single patients. Purple lines are the upper and lower limits of normal range. *P < .05 vs all groups. (C, upper panel) Experimental design. Before the experiment, HMEC-1 were left for 16 hours in medium with or without the RalA inhibitor BQU57 (10 µM). Thereafter, HMEC-1 were activated for 10 minutes with ADP, exposed for 3 hours to ctr or aHUS serum, and then perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. Formation of platelet aggregates was evaluated at the end of blood perfusion. (C, lower panel) Endothelial surface area covered by platelet aggregates. Results are shown as fold increase of stained surface area after incubation with aHUS serum (n = 3) vs ctr serum run in parallel. Data are mean ± SD. Red points represent fold increase values of single patients. Purple lines are the upper and lower limits of normal range. *P < .05 vs all groups. (D) Representative images of experiments (green staining) relative to Figure 4C. Original magnification ×200. Scale bar represents 50 μm.
To assess whether the prothrombotic effect of aHUS serum was ascribable to the exocytosis of WPBs, experiments were repeated with HMEC-1 preincubated with BQU57. BQU57 fully prevented platelet adhesion and aggregation on cells exposed to aHUS serum (Figure 4C-D, absolute values in pixel2 ; additional representative images in supplemental Figure 10).
Role of C5a/C5aR1-induced exocytosis of WPBs in COVID-19–associated thrombosis
There is growing evidence that unrestrained complement activation induced by SARS-CoV-2 infection plays a major role in endothelial cell dysfunction, thrombus formation, and intravascular coagulation often observed in patients with COVID-19.13 We evaluated whether the complement terminal pathway was activated in patients with severe COVID-19 (patients with respiratory distress requiring ventilation support; Table 2). HMEC-1 exposed to COVID-19 serum exhibited significantly higher C5b-9 formation on the cell surface than HMEC-1 incubated with ctr serum (Figure 5A). Notably, the amount of C5b-9 formed was comparable to that observed on cells exposed to aHUS serum. Plasma C5a levels in patients with COVID-19 were significantly higher than in healthy subjects, confirming terminal pathway activation in vivo as well (Figure 5B).

Terminal complement activation, and role of C5a/C5aR1 signaling in COVID-19 serum-induced platelet adhesion and aggregation on HMEC-1. (A) Endothelial surface area covered by staining for C5b-9 after incubation of ADP-activated HMEC-1 for 2 hours with ctr serum, or serum from patients with COVID-19 or aHUS, taken as positive controls. Results are shown as fold increase of stained surface area after incubation with ctr serum run in parallel. Data are mean ± SD n = 5 independent experiments. Red points represent fold increase values of single experiments. Purple lines are the upper and lower limits of normal range (calculated as shown in supplemental Figure 2). *P < .05 vs control. (B) C5a levels in plasma from healthy subjects (n = 10) or patients with COVID-19 (n = 4). Data are mean ± SD. *P < .05 vs healthy subjects. (C, upper panel) Experimental design. ADP-activated HMEC-1 were exposed for 2 hours to ctr serum or serum from patients with COVID-19 or aHUS taken as positive control, and then perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. COVID-19 serum was added or not with the C5aR1 antagonist CCX168 (200 ng/mL). Formation of platelet aggregates was evaluated at the end of blood perfusion. (C, lower panel) Endothelial surface area covered by platelet aggregates. Results are shown as fold increase of stained surface area after incubation with COVID-19 serum (n = 6) or aHUS serum (n = 4) vs ctr serum run in parallel. Data are mean ± SD. Red points represent fold increase values of single patients. Purple lines are the upper and lower limits of normal range. *P < .05 vs the groups indicated by horizontal bar. #P < .05 vs ctr serum. (D) Representative images of experiments (green staining) relative to Figure 5C. Original magnification ×200. Scale bar represents 50 μm. (E, upper panel) Experimental design. ADP-activated HMEC-1 were exposed for 2 hours to ctr serum or serum from patients with COVID-19 and then perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. COVID-19 serum was added or not with an anti-vWF antibody (10 μg/mL). Formation of platelet aggregates was evaluated at the end of blood perfusion. (E, lower panel) Endothelial surface area covered by platelet aggregates. Results are shown as fold increase of stained surface area after incubation with COVID-19 serum (n = 3) vs ctr serum run in parallel. Data are mean ± SD. Red points represent fold increase values of single patients. Purple lines are the upper and lower limits of normal range. *P < .05 vs all groups. (F, upper panel) Experimental design. Before the experiment, HMEC-1 were left for 16 hours in medium with or without the RalA inhibitor BQU57 (10 µM). Thereafter, HMEC-1 were activated for 10 minutes with ADP, exposed for 2 hours to ctr serum or COVID-19 serum, and then perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. Formation of platelet aggregates was evaluated at the end of blood perfusion. (F, lower panel) Endothelial surface area covered by platelet aggregates. Results are shown as fold increase of stained surface area after incubation with COVID-19 serum (n = 3) vs ctr serum run in parallel. Data are mean ± SD. Red points represent fold increase values of single patients. Purple lines are the upper and lower limits of normal range. *P < .05 vs all groups.
Terminal complement activation, and role of C5a/C5aR1 signaling in COVID-19 serum-induced platelet adhesion and aggregation on HMEC-1. (A) Endothelial surface area covered by staining for C5b-9 after incubation of ADP-activated HMEC-1 for 2 hours with ctr serum, or serum from patients with COVID-19 or aHUS, taken as positive controls. Results are shown as fold increase of stained surface area after incubation with ctr serum run in parallel. Data are mean ± SD n = 5 independent experiments. Red points represent fold increase values of single experiments. Purple lines are the upper and lower limits of normal range (calculated as shown in supplemental Figure 2). *P < .05 vs control. (B) C5a levels in plasma from healthy subjects (n = 10) or patients with COVID-19 (n = 4). Data are mean ± SD. *P < .05 vs healthy subjects. (C, upper panel) Experimental design. ADP-activated HMEC-1 were exposed for 2 hours to ctr serum or serum from patients with COVID-19 or aHUS taken as positive control, and then perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. COVID-19 serum was added or not with the C5aR1 antagonist CCX168 (200 ng/mL). Formation of platelet aggregates was evaluated at the end of blood perfusion. (C, lower panel) Endothelial surface area covered by platelet aggregates. Results are shown as fold increase of stained surface area after incubation with COVID-19 serum (n = 6) or aHUS serum (n = 4) vs ctr serum run in parallel. Data are mean ± SD. Red points represent fold increase values of single patients. Purple lines are the upper and lower limits of normal range. *P < .05 vs the groups indicated by horizontal bar. #P < .05 vs ctr serum. (D) Representative images of experiments (green staining) relative to Figure 5C. Original magnification ×200. Scale bar represents 50 μm. (E, upper panel) Experimental design. ADP-activated HMEC-1 were exposed for 2 hours to ctr serum or serum from patients with COVID-19 and then perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. COVID-19 serum was added or not with an anti-vWF antibody (10 μg/mL). Formation of platelet aggregates was evaluated at the end of blood perfusion. (E, lower panel) Endothelial surface area covered by platelet aggregates. Results are shown as fold increase of stained surface area after incubation with COVID-19 serum (n = 3) vs ctr serum run in parallel. Data are mean ± SD. Red points represent fold increase values of single patients. Purple lines are the upper and lower limits of normal range. *P < .05 vs all groups. (F, upper panel) Experimental design. Before the experiment, HMEC-1 were left for 16 hours in medium with or without the RalA inhibitor BQU57 (10 µM). Thereafter, HMEC-1 were activated for 10 minutes with ADP, exposed for 2 hours to ctr serum or COVID-19 serum, and then perfused for 3 minutes with heparinized whole blood, added with mepacrine, from healthy subjects. Formation of platelet aggregates was evaluated at the end of blood perfusion. (F, lower panel) Endothelial surface area covered by platelet aggregates. Results are shown as fold increase of stained surface area after incubation with COVID-19 serum (n = 3) vs ctr serum run in parallel. Data are mean ± SD. Red points represent fold increase values of single patients. Purple lines are the upper and lower limits of normal range. *P < .05 vs all groups.
Perfusion with whole blood (Figure 5C) on HMEC-1 preexposed to COVID-19 serum resulted in massive platelet adhesion and aggregation so that the endothelial cell area covered by platelet aggregates was ∼5-fold greater than on cells exposed to ctr serum and comparable to that induced by aHUS serum (Figure 5C-D, absolute values in pixel2 ; additional representative images in supplemental Figure 11). The addition of the C5a receptor antagonist CCX168 to COVID-19 serum fully prevented platelet adhesion and aggregation (Figure 5C-D).
The role of vWF and WPB exocytosis was assessed by repeating experiments in the presence of an anti-vWF antibody or preincubating HMEC-1 with BQU57, respectively. Both the anti-vWF antibody and BQU57 significantly reduced the area covered by platelet aggregates on HMEC-1 exposed to COVID-19 serum (Figure 5E-F, absolute values in pixel2 ; representative images in supplemental Figure 11).
Discussion
We demonstrated that C5a/C5aR1 interaction, by inducing the exocytosis of WPBs from endothelial cells, mediates endothelial dysfunction and prothrombotic transformation in different pathological conditions associated with activation of the complement terminal pathway, from rare diseases determined by genetic dysregulations of the alternative pathway to infectious diseases.
The activation of the terminal pathway leads to the formation of C5a and C5b-9. Both the binding of C5a to C5aR1, and the formation of sublytic amounts of C5b-9, can cause profound perturbations of the physiologically thromboresistant phenotype of endothelial cells, including upregulation of tissue factor and adhesion molecules, loss of anticoagulant heparan sulfate proteoglycans, and exocytosis of P-selectin and vWF.22-28
Here, through in vitro studies with serum and cultured microvascular endothelial cells, we document that in patients with aHUS, a prototypical complement-related rare disease characterized by microvascular thrombosis, both terminal pathway effectors are produced in excessive amounts at the endothelial cell level. Our findings that blocking the C5a/C5aR1 interaction, rather than forming C5b-9, fully prevented the prothrombogenic effect of aHUS serum on endothelial cells suggest C5a/C5aR1 signaling as a major player of microvascular thrombosis in aHUS.
This possibility is supported by our data that treating patients with aHUS with the C5aR1 antagonist CCX16816 normalized excessive serum-induced platelet adhesion and aggregation ex vivo.
It is well known that patients with aHUS suffer from complement alternative pathway dysregulation due to genetic or autoimmune abnormalities.6,11 The finding here that the addition of C5a to serum from healthy subjects is enough to fully recapitulate the prothrombotic effect of aHUS serum indicates that C5a/C5aR1 interaction on the endothelium may favor platelet adhesion and aggregation, even in the absence of abnormalities in complement alternative pathway regulation. Based on these data, we hypothesize that C5a/C5aR1 signaling is a common mechanism of endothelial thromboresistance loss in conditions associated with excessive C5a production, such as those triggered by environmental factors such as infections, or tissue damage, such as ischemia reperfusion injury.29-31
In this study, we also wanted to elucidate the events induced on endothelial cells by C5a/C5aR1 interaction to understand the mechanism(s) leading to endothelial alterations. C5aR1 is a G protein–coupled receptor whose engagement activates different biological processes, including cytoskeletal reorganization and exocytosis.32,33 On endothelial cells, C5a/C5aR1 signaling induces the formation of actin stress fibers that cause cytoskeletal changes and, eventually, cell retraction and loss of endothelial barrier integrity.34 In addition, C5aR1 engagement in blood leukocytes stimulated exocytosis of intracellular vesicles. In fact, binding of C5a to C5aR1 on mast cells and granulocytes led to exocytosis of the intracellular granules containing histamine.35,36 Our results, which show that exposure of endothelial cells to C5a induced rapid release of vWF and P-selectin (the main molecular components of the endothelial secretory vesicles WPBs), indicate that C5aR1 activates the exocytosis pathway in the endothelium as well. This possibility is confirmed by the finding that a specific inhibitor of the small GTPase RalA, which is central to the molecular machinery that guides WPB exocytosis,19-21 normalized vWF staining on C5a-stimulated endothelial cells.
Both P-selectin and vWF expressed on activated endothelial cells can recruit circulating vWF. Indeed, an earlier study has shown that the luminal domain of P-selectin strongly binds the D′-D3 vWF domains.37 In addition, Ulrichts et al have demonstrated that soluble vWF associates with immobilized vWF through multiple domain interactions, thus resulting in an adhesive scaffold.38 Consistent with this finding, here we demonstrate that following C5a-induced exocytosis of P-selectin and vWF on HMEC-1, exposure to serum resulted in high amounts of circulating vWF gathering on the endothelial cell surface.
vWF is a large multimeric adhesive protein that mediates platelet adhesion to the endothelium and platelet thrombus formation.39 The key role of vWF in mediating C5a-induced prothrombogenic action was demonstrated here by results showing that formation of platelet aggregates on endothelial cells preexposed to C5a and normal human serum was fully prevented by neutralizing vWF. Similarly, either blocking WPB exocytosis or neutralizing vWF fully prevented the formation of platelet aggregates induced by aHUS serum on HMEC-1, indicating that C5a-mediated WPB exocytosis and the gathering of circulating vWF on the endothelial cell surface may play a role in aHUS-associated microvascular thrombosis.
Once we established that C5a induced endothelial dysfunction through exocytosis of WPBs in patients with genetically determined complement dysregulation, we investigated whether such mechanisms also exist in individuals without genetic complement abnormalities who suffered from acute activation of complement by strong environmental triggers such as severe viral infections.
To address this goal, we focused on patients with severe COVID-19 who share both complement activation and microvascular injury with aHUS.13,40 SARS-CoV-2 viral invasion triggers an uncontrolled immunological response that includes unrestrained activation of all three complement pathways13,41,42 until the terminal step. This finding is documented by elevated levels of sC5b-9 and C5a in the plasma of patients with severe COVID-1943-45 (and present data).
Extensive endotheliitis has been observed in the lungs and other organs of patients with severe COVID-19.46-48 These lesions were associated with microvascular injury as well as thrombosis of the medium and large vessels, which can cause pulmonary embolism.49,50 The above data, along with elevated plasma concentrations of vWF and P-selectin in patients with severe COVID-19–associated coagulopathy,51 suggest massive endothelial cell activation leading to the release of WPBs.
Our finding that serum from patients with COVID-19 on CPAP induced massive formation of C5b-9 on HMEC-1 indicates that the endothelium is a target of terminal complement products in severe COVID-19. This finding is in line with published studies showing that in lung sections of deceased patients with COVID-19, deposits of C5b-9 colocalized with endotheliitis lesions.44,49 It is relevant here that we have shown that serum from patients with COVID-19 has a strong prothrombotic effect on endothelial cells, which was completely blocked by inhibiting C5aR1, as well as by preventing WPB exocytosis. Elevated interleukin-6 levels, which are a common finding in patients with COVID-19,52 could contribute to upregulate C5aR in endothelial cells.53 We therefore propose that the C5a/C5aR1 axis plays a role in the development of vascular inflammation, endothelial dysfunction, and thrombosis in COVID-19.
In summary, the results presented in this article from two prototypical conditions characterized by either genetically or environmentally determined terminal pathway activation indicate that C5a/C5aR1 signaling is a pathogenetic driver of endothelial dysfunction and thrombosis. We also propose that exocytosis of WPBs with vWF release and plasma vWF recruitment on endothelial cells is a mechanistic explanation for the prothrombogenic effects of C5a/C5aR1 (as summarized in Figure 6). The pathogenetic analogies we found between aHUS and COVID-19–associated thrombosis make C5a/C5aR1 a candidate mediator for other thrombotic and coagulation disorders, including rare severe cerebral venous thrombosis episodes that were recently brought to the public’s attention.54
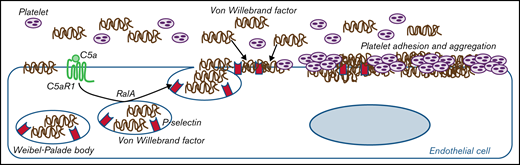
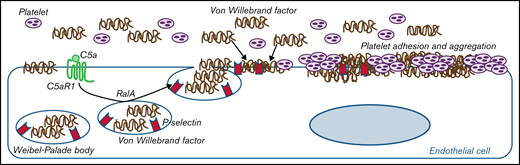
Proposed mechanism underlying the pro-thrombotic action of C5a/C5aR1 signaling. Upon engagement with its receptor C5aR1, C5a induces the activation of RalA, which leads to the exocytosis of Weibel-Palade bodies (WPB). Then, vWF and P-selectin are exocytosed from WPBs, providing an adhesive scaffold on which circulating vWF is in turn recruited. As a consequence for the above events, massive platelet adhesion and aggregation occur on the endothelial cell surface.
Proposed mechanism underlying the pro-thrombotic action of C5a/C5aR1 signaling. Upon engagement with its receptor C5aR1, C5a induces the activation of RalA, which leads to the exocytosis of Weibel-Palade bodies (WPB). Then, vWF and P-selectin are exocytosed from WPBs, providing an adhesive scaffold on which circulating vWF is in turn recruited. As a consequence for the above events, massive platelet adhesion and aggregation occur on the endothelial cell surface.
These findings may have important clinical implications. In trials and clinical practice, blocking the complement terminal pathway with eculizumab protected patients with aHUS from microvascular thrombosis6 and has become the therapy of choice for this condition. Published case series and proof-of-concept studies have described patients with severe COVID-19 who received off-label treatment with eculizumab. Although there were variable degrees of efficacy, possibly depending on time of drug administration, dose and disease severity, a subgroup of patients with COVID-19 treated with eculizumab exhibited improvements in respiratory symptoms and pulmonary lesions as well as markers of systemic inflammation.43,55,56 Notably, treatment with eculizumab reduced systemic hypercoagulation, as shown by decreased circulating levels of d-dimer compared with values before treatment.57
Based on these studies, several randomized controlled trials have been started. However, interim analysis of a large phase III trial with the anti-C5 antibody ravulizumab (#NCT04369469) in patients with COVID-19 already on mechanical ventilation at randomization could not demonstrate clinical benefit leading to a study arrest. It is likely that in advanced disease, the hyperinflammation state is driven by several pathways, and targeting only one of them is not enough to reverse disease. In this regard, in a small study of 17 consecutive patients with COVID-19 with acute respiratory distress, patients (n = 7) treated with eculizumab combined with a JAK1/2 inhibitor to block cytokine signaling58 showed significant improvements in respiratory symptoms and radiographic pulmonary lesions compared with patients (n = 10) receiving the best supportive care only.58
In the ZILUCOV trial (#NCT04382755), 81 patients with COVID-19 with signs of hypoxia not yet requiring mechanical ventilation have been randomized to receive the C5 antagonist zilucoplan or standard-of-care treatment.59 The trial has completed recruitment, and it is hoped that results will clarify whether early intervention with C5 blockade could improve short- and long-term outcomes of patients with COVID-19.
Selective blocking of C5a/C5aR1 could have advantages over C5 blockade. Indeed, blocking C5a/C5aR1 leaves C5b intact and preserves the formation of the C5b-9 membrane attack complex, which plays a key role in controlling several bacterial infections that often develop as comorbid conditions in these patients. In addition, this approach will inhibit the C5a-mediated recruitment of neutrophils and monocytes in damaged organs44 and limit the production of inflammatory molecules as well as prothrombotic factors from immune cells, platelets, and endothelium.13,29,60-63 A potential beneficial effect of C5a inhibition in COVID-19 is suggested by secondary outcome results from an exploratory randomized phase 2 trial involving patients with severe COVID-19, which showed a lower incidence of serious pulmonary embolisms in patients receiving the anti-C5a antibody IFX-1 than in the control group receiving best supportive care only.64 However, the study was not powered on this end point, and results should be confirmed in the ongoing large, multicenter, placebo-controlled trial involving 390 patients (#NCT04333420).
A previous study using a murine model of aHUS generated by introducing the p.W1206R mutation in mouse FH (FHR/R mice) showed that C5aR1 genetic deficiency prevented macrovessel thrombosis in FHR/R mice, whereas C6 or C9 deletion, which prevented C5b-9 formation, diminished renal thrombotic microangiopathy, indicating that C5aR1 and C5b-9 play different pathogenetic roles in this model.14 However, the FHR/R mouse model does not fully recapitulate the human aHUS phenotype because the animals developed systemic thrombophilia, and thrombocytopenia and anemia persisted in FHR/RC6−/− mice, in which renal thrombotic microangiopathy was prevented.14 Finally, mouse and human complement systems differ particularly in the type, function, and distribution of regulatory proteins.65
Both C5a22 and C5b-9 have been shown to induce tissue factor activity on endothelial cells22,66,67 and may synergize in inducing a prothrombotic phenotype. Further studies are also required to assess the role of C5b-9 per se and of C5b-9–mediated hemolysis (and subsequent heme-mediated kidney toxicity) in kidney injury in patients with aHUS and COVID-19.68,69
The potential therapeutic benefit of blocking the downstream signaling activated by C5a/C5aR1, such as neutralizing vWF or preventing exocytosis of WPBs through specific inhibition of RalA activity, a target already under study for cancer therapy,70,71 is worth investigating as well.
Acknowledgments
The authors are grateful to Silvia Prandini, Diana Cadè, Veruscka Lecchi, Erica Daina, and Elena Bresin for their precious support in organizing and managing sample and clinical data collection for in vitro experiments involving patients and healthy subjects. The authors thank Ilian Iliev, Carolina Aparicio, and Matias Trillini for following the patients during the ACCESS Study. The authors also wish to thank Paolo Macor for providing the anti-C7 antibody and the blocking human complement C5 minibody and performing C5a measurement in serum incubated with HMEC-1. The authors also are grateful to Kerstin Mierke for editing the manuscript and Manuela Passera for providing secretarial assistance.
This work was partially supported by the Italian Ministero della Salute (RF-2016-02361720). L.L. is a recipient of a fellowship from Fondazione Aiuti per la Ricerca sulle Malattie Rare ARMR ONLUS (Bergamo, Italy).
Authorship
Contribution: S.A., M.G., S. Gastoldi, and M.N. designed the study; S. Gastoldi and D.S. conducted immunofluorescence experiments; S. Gastoldi conducted platelet adhesion and aggregation experiments; L.L. performed genetic analysis; S.C. and M.T. performed analysis of platelet aggregates by scanning electron microscopy; S.A., M.G., S. Gastoldi, and M.N. analyzed and interpreted the experimental results; S.A. wrote the first draft of the manuscript revised with M.N. and M.G. P.R., V.P., S.R., S. Gamba, and N.R. organized, managed, and monitored clinical studies and collected patient samples; and A.B. and G.R. revised the manuscript and provided critical discussion.
Conflict-of-interest disclosure: M.N. received honoraria from Alexion Pharmaceuticals for giving lectures and participating in advisory boards and research grants from Omeros, Chemocentryx, Inception Science Canada, BioCryst Pharmaceuticals, Novartis, and Roche. A.B. received honoraria from Boehringer Inghelheim, Alexion Phamaceuticals, Janssen Pharmaceuticals, Akebia Therapeutics, Inception Science Canada, and BioCryst Pharmaceuticals. G.R. has consultancy agreements with Boehringer Inghelheim, Janssen Pharmaceuticals, Akebia Therapeutics, Alexion Pharmaceuticals, Alnylam, Inception Science Canada, and BioCryst Pharmaceuticals. M.G. has received research grants from Omeros, Novartis, and Roche. The remaining authors declare no competing financial interests.
The funding sources had no role in study design or collection, analysis, and interpretation of data nor in the writing of the report or the decision to submit the paper for publication.
Correspondence: Marina Noris, Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Via Camozzi 3, 24020 Bergamo, Italy; e-mail: marina.noris@marionegri.it.