Key Points
A patient-derived iPSC model recapitulates defective erythroid maturation in female XLSA.
Azacitidine reactivates the silent wild-type ALAS2 allele and ameliorates inefficient erythropoiesis in iPSC-derived HPCs from female XLSA.
Abstract
X-linked sideroblastic anemia (XLSA) is associated with mutations in the erythroid-specific δ-aminolevulinic acid synthase (ALAS2) gene. Treatment of XLSA is mainly supportive, except in patients who are pyridoxine responsive. Female XLSA often represents a late onset of severe anemia, mostly related to the acquired skewing of X chromosome inactivation. In this study, we successfully generated active wild-type and mutant ALAS2-induced pluripotent stem cell (iPSC) lines from the peripheral blood cells of an affected mother and 2 daughters in a family with pyridoxine-resistant XLSA related to a heterozygous ALAS2 missense mutation (R227C). The erythroid differentiation potential was severely impaired in active mutant iPSC lines compared with that in active wild-type iPSC lines. Most of the active mutant iPSC-derived erythroblasts revealed an immature morphological phenotype, and some showed dysplasia and perinuclear iron deposits. In addition, globin and HO-1 expression and heme biosynthesis in active mutant erythroblasts were severely impaired compared with that in active wild-type erythroblasts. Furthermore, genes associated with erythroblast maturation and karyopyknosis showed significantly reduced expression in active mutant erythroblasts, recapitulating the maturation defects. Notably, the erythroid differentiation ability and hemoglobin expression of active mutant iPSC-derived hematopoietic progenitor cells (HPCs) were improved by the administration of δ-aminolevulinic acid, verifying the suitability of the cells for drug testing. Administration of a DNA demethylating agent, azacitidine, reactivated the silent, wild-type ALAS2 allele in active mutant HPCs and ameliorated the erythroid differentiation defects, suggesting that azacitidine is a potential novel therapeutic drug for female XLSA. Our patient-specific iPSC platform provides novel biological and therapeutic insights for XLSA.

Introduction
Congenital sideroblastic anemias are a group of heterogeneous inherited diseases characterized by impaired heme synthesis and mitochondrial iron overload with ring sideroblasts in the bone marrow. X-linked sideroblastic anemia (XLSA) is the most common hereditary sideroblastic anemia and is caused by mutations in the erythroid-specific δ-aminolevulinic acid synthase (ALAS2) gene, which is the first enzyme involved in heme biosynthesis in erythroid cells.1,2 Nearly 100 different ALAS2 mutations have been reported to date.3 XLSA occurs preferentially in males and usually presents as asymptomatic or mild microcytic anemia. However, approximately one-fourth of the affected XLSA probands are females, and those patients show a distinct clinical presentation and mutation spectrum.4-6 Unlike males, females with XLSA often harbor heterozygous deleterious ALAS2 mutations that are embryonic lethal in males and develop more severe clinical phenotypes with macrocytic anemia in mid-to-late adulthood.7 Their anemia is mostly associated with skewed X-chromosome inactivation, which increases with age.8,9 Most families with female XLSA have multiple affected members, indicating a familial predisposition to acquired X-chromosome skewing.3
One treatment option is pyridoxine, which metabolizes as pyridoxal 5′-phosphate, the cofactor for ALAS2. However, pyridoxine refractoriness is reported in nearly half of cases, including those with an R227C mutation.1,10 The severe loss of function caused by this mutation may explain why no male carriers have been identified in affected families.11 Therapies for XLSA in patients who do not respond to pyridoxine are mostly aimed at supportive care. Administration of δ-aminolevulinic acid (ALA) could ameliorate the heme biosynthesis defects that occur in XLSA, because ALAS2 catalyzes the condensation of glycine and succinyl-CoA into ALA. However, a recent study of a single case showed no improvement in the hemoglobin level even at the in vitro effective concentration.12 Therefore, other treatment approaches should be explored.
Several cell and animal models of male XLSA have been developedb13-18 but there are no reported models of female XLSA with heterozygous ALAS2 mutations. In this study, we established active wild-type induced pluripotent stem cell (WT iPSC) and active-mutant induced pluripotent stem cell (MT iPSC) lines from the peripheral blood cells of an affected mother and her 2 daughters in a family with XLSA caused by a heterozygous ALAS2-R227C mutation. We then differentiated the iPSCs into erythroblasts to model the pathophysiology of female XLSA. Using this iPSC-based platform, we found that the demethylating agent azacitidine (AZA) induces the expression of WT ALAS2 in active mutant iPSC-derived hematopoietic progenitor cells (MT HPCs) to ameliorate abnormal erythroid differentiation caused by the heme biosynthesis defects.
Materials and methods
Generation of iPSCs from patients with XLSA
This study was approved by the Ethics Committees of the Graduate School of Medicine, Kyoto University, and Kyoto University Hospital. Written informed consent was obtained from all patients, and the study was performed in accordance with the Declaration of Helsinki. XLSA-iPSCs were established from 3 patients harboring a heterozygous R227C mutation using episomal vectors, as previously described.19 In brief, the patients’ peripheral blood mononuclear cells (PBMCs) were transfected with episomal vector mixtures including pCXLE-hOCT3/4-shp53, pCXLE-hSK, pCXLE-hUL, and pCXWB-EBNA1, and then seeded onto mouse embryonic fibroblasts feeder cells. The iPSC IDs of all XLSA-iPSC lines used in the differentiation experiment are described in supplemental Table 1. The WT and MT iPSC lines derived from SA1 are denoted as WT1-iPSC1, -2, and -3 and MT1-iPSC1, -2, -3, and -4, respectively. Similarly, those from SA3 are denoted as WT3-iPSC1 and -2 and MT3-iPSC1 and -2, respectively. We used an iPSC line derived from a healthy donor (692D2) as the control.19 Chromosomal G-band analysis of the XLSA-iPSCs was performed at the Nihon Gene Research Laboratories (Sendai, Japan). Animal experiments for the teratoma formation of XLSA-iPSCs were performed as previously described, in accordance with the code of ethics of the Animal Research Committee of Kyoto University.20
Human androgen receptor assay and Sanger sequencing
Genomic DNAs from iPSCs, CD43+CD34+ iPSC–derived HPCs, and MACS–sorted CD34+ and CD235a+ bone marrow cells were extracted for the HUMARA assay using the DNeasy Blood and Tissue kit (Qiagen). DNA samples (200 ng) were incubated overnight at 37°C in 20-μL reactions containing 10 U HhaI and 10 U RsaI. The digested DNA samples were purified on QIAquick spin columns (Qiagen) and amplified with a Veriti Thermal Cycler (ThermoFisher Scientific). Polymerase chain reactions (PCRs) and analyses of the PCR products were performed as previously described.21,22 Peripheral red blood cells were separated with Polymorphprep (Abbott Diagnostics Technologies AS), according to the manufacturer’s protocol. Sanger sequencing of exon 6 of the ALAS2 gene was performed as previously reported.6
Hematopoietic differentiation and erythroid differentiation assays
The hematopoietic differentiation of iPSCs was performed with embryoid body (EB) formation, and feeder-free erythroid differentiation was performed as previously reported.23,24 For feeder-free erythroid differentiation with AZA, FACS-sorted, iPSC-derived HPCs on day 15 were differentiated in StemPro-34 medium (Invitrogen). The cytokines used for the differentiation were as follows: on days 15 to 23, human stem cell factor (hSCF, 100 ng/mL; R&D Systems), human erythropoietin (hEPO, 4 U/mL; Kyowa Kirin Co, Ltd.), and human interleukin-3 (hIL-3; 5 ng/mL, R&D Systems); on days 23 to 26, hSCF (100 ng/mL) and hEPO (4 U/mL); and on days 26 to 29, hEPO (4 U/mL). Erythroid differentiation on OP9 stromal feeder cells was performed as previously described with some modifications.16,18,25 In brief, FACS-sorted HPCs on day 15 were plated onto OP9 cells and cultured and maintained in α-minimum essential medium (Invitrogen) supplemented with 10% fetal calf serum, 100 mM glutamine (Invitrogen), 100 μM monothioglycerol (Sigma Aldrich), 5 μg/mL transferrin (Roche), and 50 μg/mL ascorbic acid (Sigma Aldrich). The cytokines and chemical compounds used during the differentiation were as follows: on days 15 to 20, hSCF (100 ng/mL), human Fms-like tyrosine kinase 3 ligand (hFlt-3; 100 ng/mL, R&D Systems), hEPO (4 U/mL), and human thrombopoietin (hTPO; 50 ng/mL R&D Systems); on days 20 to 27, hSCF (100 ng/mL) and hEPO (4 U/mL); and on days 27 to 34, hEPO (4 U/mL) and sodium ferrous citrate (SFC; 250 μM, Nihon Generic Co, Ltd).
Treatment with ALA and AZA
From the beginning of erythroid differentiation, 500 μM ALA (Sigma) or 100 or 500 nM AZA (Nacalai Tesque) was added to the cultures. For the ALA-treated cells, ALA was added daily to the erythroid differentiation medium. For the AZA-treated cells, AZA was added at the time of the change of medium (in the EB-feeder–free method, on days 19, 23, and 26; in the EB-OP9 method, on days 23 and 27). For the methylcellulose assay, AZA was added to the medium at the beginning of the colony-forming unit (CFU) assay.26
Statistical analyses
GraphPad Prism 9 (GraphPad Software) was used to perform all statistical analyses. P < .05 indicated significant results.
Results
Female patients with late-onset XLSA show preferential inactivation of the paternal X chromosome at the stage of hematopoietic progenitor cells
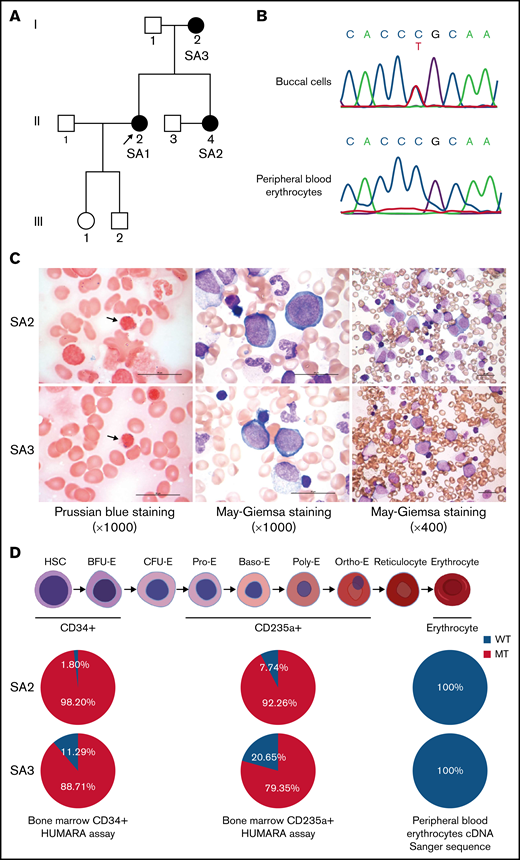
We had identified a family with XLSA harboring a heterozygous ALAS2-R227C mutation (c.679C>T, NCBI NM_000032.4; Figure 1A).27 The clinical features of the proband (SA1) and her mother (SA3), who were diagnosed with XLSA caused by the mutation, have been published.6 Her younger sister (SA2) also presented late-onset severe macrocytic anemia and evidence of iron overload (Table 1). Sanger sequencing of the genomic DNA from SA2 buccal cells demonstrated the same heterozygous ALAS2 missense mutation as the proband. We also checked the ALAS2 complementary DNA sequence of her peripheral blood erythrocytes and found no ALAS2 mutation (Figure 1B). The bone marrow aspirate showed erythroid hyperplasia with ring sideroblasts and dyserythropoiesis, such as megaloblastic changes, nuclear budding, and karyorrhexis, but no apparent abnormalities of megakaryocytes and granulocytes (Figure 1C). Although the CD34+ and CD235a+ bone marrow cells of SA2 revealed apparent skewing toward the mutant ALAS2 allele, the peripheral blood erythrocytes expressed only WT ALAS2 (Figure 1D). Her condition was diagnosed as XLSA. We also obtained bone marrow aspirate from SA3, and it verified similar erythroid-restricted dysplasia (Figure 1C-D). The clinical characteristics of the patients are shown in Table 1, including laboratory data at the time of diagnosis. SA1 had received 60 mg pyridoxal phosphate hydrate for 3 months with no improvement in her anemia (supplemental Figure 1). Therefore, a clinical trial of ALA administration was performed with the consent of SA1 and SA2. They received 150 mg of ALA-phosphate (kindly provided by SBI Pharmaceuticals, Tokyo, Japan) for 12 and 24 weeks. There were no side effects of ALA administration; however, the anemia did not improve (supplemental Figure 2).
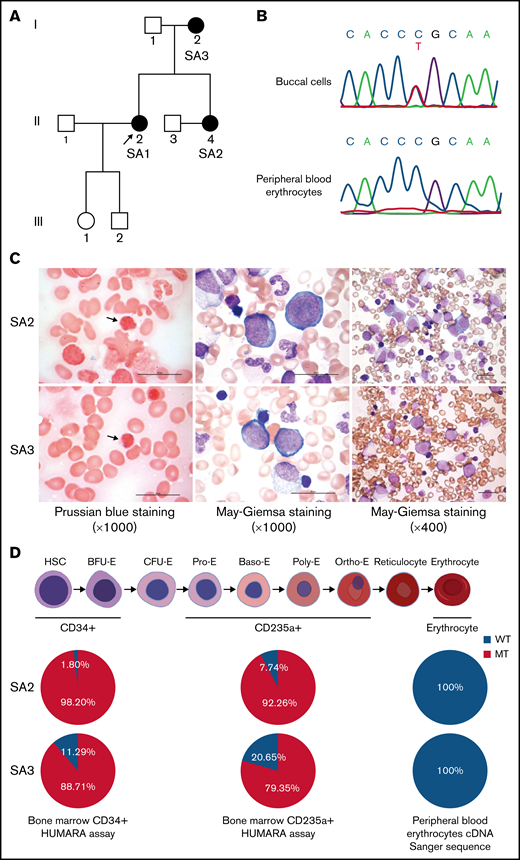
Identification of XLSA in a family harboring the heterozygous ALAS2-R227C mutation. (A) Pedigree of the family. Squares indicate males, and circles indicate females. Filled circles denote the patients confirmed by ALAS2 sequencing. The arrow indicates the proband. (B) Sanger sequencing data of ALAS2 genomic DNA from SA2 buccal cells (top) and ALAS2 cDNA from SA2 peripheral blood erythrocytes (bottom) are shown. (C) Morphology of SA2 and SA3 bone marrow cells. An increased number of ring sideroblasts was observed with Prussian blue staining (left). The arrows indicate ring sideroblasts. Megaloblastic change was detected by May Grunwald-Giemsa staining (middle and right). Magnification of the objective lens: ×100 (left and middle) and ×40 (right). Bars represent 25 μm. (D) Summary of HUMARA assays of CD34+ and CD235a+ bone marrow cells and ALAS2 cDNA Sanger sequencing of peripheral blood erythrocytes. The schematic diagram of erythropoiesis was created using BioRender.com. cDNA, complementary DNA.
Identification of XLSA in a family harboring the heterozygous ALAS2-R227C mutation. (A) Pedigree of the family. Squares indicate males, and circles indicate females. Filled circles denote the patients confirmed by ALAS2 sequencing. The arrow indicates the proband. (B) Sanger sequencing data of ALAS2 genomic DNA from SA2 buccal cells (top) and ALAS2 cDNA from SA2 peripheral blood erythrocytes (bottom) are shown. (C) Morphology of SA2 and SA3 bone marrow cells. An increased number of ring sideroblasts was observed with Prussian blue staining (left). The arrows indicate ring sideroblasts. Megaloblastic change was detected by May Grunwald-Giemsa staining (middle and right). Magnification of the objective lens: ×100 (left and middle) and ×40 (right). Bars represent 25 μm. (D) Summary of HUMARA assays of CD34+ and CD235a+ bone marrow cells and ALAS2 cDNA Sanger sequencing of peripheral blood erythrocytes. The schematic diagram of erythropoiesis was created using BioRender.com. cDNA, complementary DNA.
Generation of MT and isogenic WT iPSC lines from female patients with XLSA
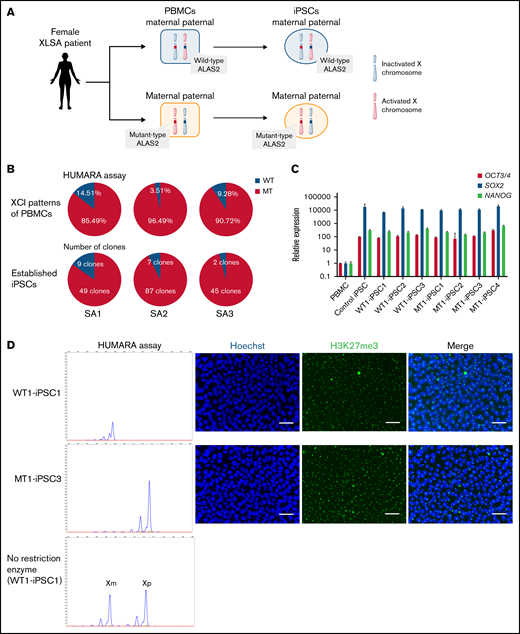
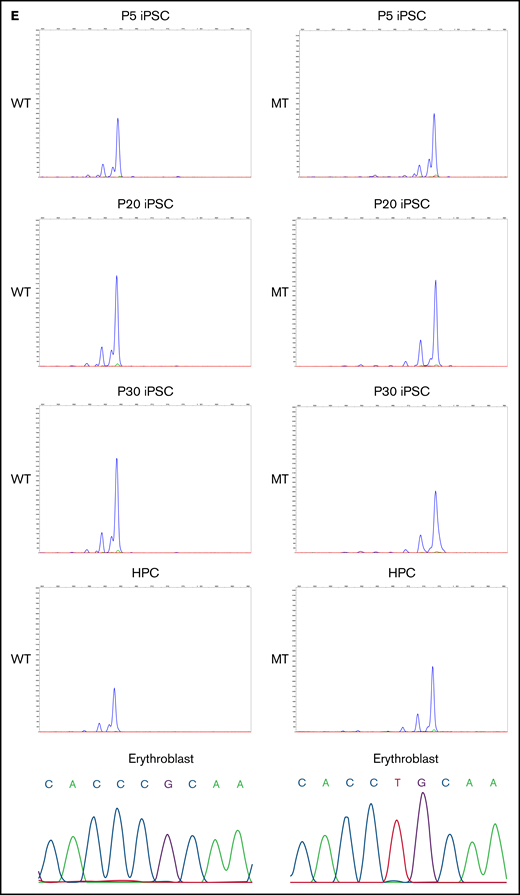
We generated iPSCs from the peripheral blood cells of all 3 patients. Because the ALAS2 gene is located on the X chromosome, 1 of the 2 ALAS2 genes is randomly inactivated by lyonization in female patients. Therefore, the cells in which the X chromosome with mutated ALAS2 is inactivated (WT cells) and the cells in which the X chromosome with normal ALAS2 is inactivated (MT cells) were established from the same patient (Figure 2A; supplemental Table 1). We generated 58 iPSC lines from SA1, 94 iPSC lines from SA2, and 47 iPSC lines from SA3. HUMARA assays demonstrated that the PBMCs of all 3 patients underwent skewed X-chromosome inactivation with preferential inactivation of the X chromosome with WT ALAS2 (Figure 2B). The percentage of MT iPSC lines was similar to the percentage of X-chromosome inactivation in PBMCs for all patients with XLSA (Figure 2B). The gene expression of pluripotent markers OCT3/4, SOX2, and NANOG was verified in 3 WT and 4 MT iPSC lines derived from SA1 according to quantitative reverse transcription-PCR (qRT-PCR) analysis performed with the primers in supplemental Table 2 (Figure 2C). These 7 iPSC lines expressed the pluripotent markers SSEA-4 and TRA1-60 and formed teratomas in vivo (supplemental Figure 3A-B). Karyotype analysis revealed no abnormalities (supplemental Figure 3C). Using the HUMARA assay and immunostaining of H3K27me3, we demonstrated that the maternal X chromosome of the WT iPSC lines was inactivated, whereas the paternal X chromosome of the MT iPSC lines was inactivated (Figure 2D). The X-chromosome inactivation status was retained in the iPSC lines after passaging. The maternal X chromosome of the WT iPSC lines, which was inactivated immediately after iPSC establishment (passage 5), remained inactivated at passages 20 and 30. The paternal X chromosome of the MT iPSC lines also retained the inactivated state after repeated passages. Notably, X-chromosome inactivation was maintained after hematopoietic differentiation and further differentiation into erythroblasts (Figure 2E).

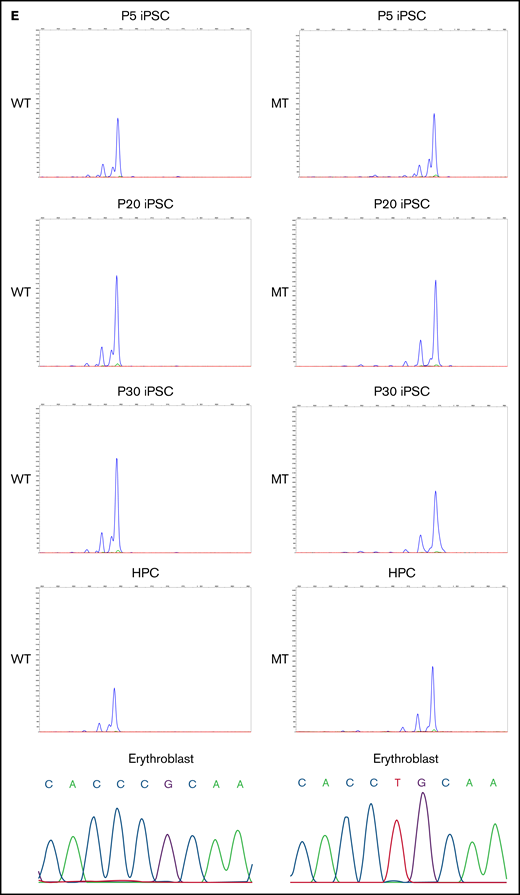
Generation of XLSA patient-derived iPSCs and confirmation of X-chromosome inactivation. (A) Schematic representation of the iPSC generation from PBMCs of female patients with XLSA who harbored a heterozygous ALAS2 mutation. The figure was created using BioRender.com. (B) Summary of HUMARA assays of PBMCs from patients with XLSA and the percentage of MT iPSC lines among established iPSCs. (C) Expression levels of OCT3/4, SOX2, and NANOG genes in PBMCs, 1 control iPSC line derived from a healthy donor, 3 WT iPSC lines, and 4 MT iPSC lines. Expression levels of PBMCs were set to 1. Each line was tested in 3 independent experiments. (D) HUMARA assays of WT and MT iPSCs (left). The undigested control is shown at the bottom left. Representative immunofluorescence staining images for Hoechst expression and H3K27me3 expression in iPSCs derived from patients with XLSA (right). Magnification of the objective lens: ×20. Bars represent 50 μm. (E) HUMARA assays of representative iPSC lines (WT1-iPSC3 and MT1-iPSC2) after 5, 20, and 30 passages and HPCs derived from the iPSC lines. Complementary DNA Sanger sequencing data of erythroblasts derived from the iPSC lines are shown on the far right.
Generation of XLSA patient-derived iPSCs and confirmation of X-chromosome inactivation. (A) Schematic representation of the iPSC generation from PBMCs of female patients with XLSA who harbored a heterozygous ALAS2 mutation. The figure was created using BioRender.com. (B) Summary of HUMARA assays of PBMCs from patients with XLSA and the percentage of MT iPSC lines among established iPSCs. (C) Expression levels of OCT3/4, SOX2, and NANOG genes in PBMCs, 1 control iPSC line derived from a healthy donor, 3 WT iPSC lines, and 4 MT iPSC lines. Expression levels of PBMCs were set to 1. Each line was tested in 3 independent experiments. (D) HUMARA assays of WT and MT iPSCs (left). The undigested control is shown at the bottom left. Representative immunofluorescence staining images for Hoechst expression and H3K27me3 expression in iPSCs derived from patients with XLSA (right). Magnification of the objective lens: ×20. Bars represent 50 μm. (E) HUMARA assays of representative iPSC lines (WT1-iPSC3 and MT1-iPSC2) after 5, 20, and 30 passages and HPCs derived from the iPSC lines. Complementary DNA Sanger sequencing data of erythroblasts derived from the iPSC lines are shown on the far right.
Erythroid differentiation is impaired in MT iPSCs
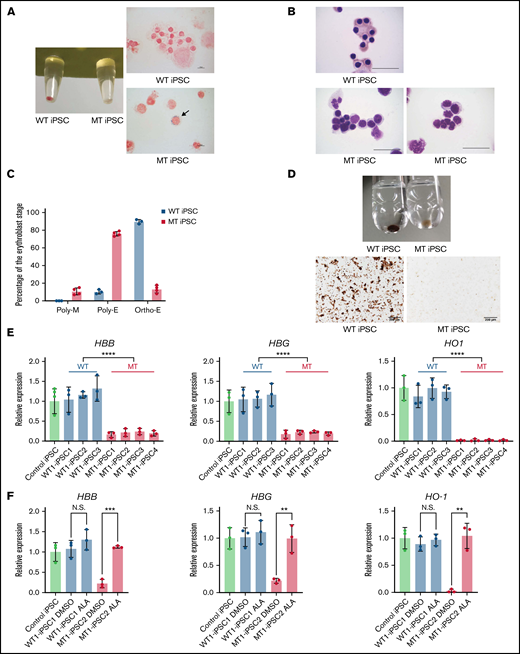
Next, we differentiated XLSA-iPSCs into HPCs by EB formation. The hematopoietic maturation capacity was assessed by CFU assays in iPSC-derived HPCs from SA1. No difference was observed in the total colony count between the 2 types of HPCs (Figure 3A). Although erythroid and mixed colonies were observed in the CFU assay of WT iPSC lines, no erythroid colonies and only a few mixed colonies were observed in the MT iPSC lines (Figure 3B-C). Lentiviral transduction of WT ALAS2 improved erythroid colony formation in MT HPCs (Figure 3D; supplemental Figure 4A). We further investigated the erythroid differentiation defects in MT iPSC lines using a feeder-free erythroid culture system. Hematopoietic differentiation was comparable between the 2 types of iPSC lines (Figure 3E). However, further differentiation into CD235+ erythroid cells was significantly impaired in the MT iPSC lines compared with that in the WT iPSC lines derived from both SA1 and SA3 (Figure 3F; supplemental Figure 4B-C). In our feeder-free erythroid differentiation method, most MT HPCs died at an early stage of differentiation (supplemental Figure 4D). As a result, we could neither identify morphological characteristics, such as ring sideroblasts or megaloblastic changes, nor perform a precise gene expression analysis of the erythroblasts expressing mutant ALAS2. Therefore, we designed a coculture system with OP9 feeder cells to successfully recapitulate the morphological features of mutant ALAS2-expressing erythroblasts. In this system, most MT HPCs survived for extended periods (supplemental Figure 4E-F). The pellet of active WT iPSC–derived erythroblasts (WT erythroblasts) was red, whereas that of active mutant iPSC-derived erythroblasts (MT erythroblasts) was white (Figure 4A). Iron staining revealed ring sideroblasts in MT erythroblasts, but not in WT erythroblasts (Figure 4A). May Grunwald-Giemsa staining showed that most WT erythroblasts were mature, whereas most MT erythroblasts exhibited immature morphological phenotypes. In addition, dysplastic changes, such as nuclear budding and karyorrhexis, were observed in MT erythroblasts, similar to the primary erythroblasts of the patient (Figure 4B). Although most of the WT erythroblasts were orthochromatic, most of the MT erythroblasts were polychromatic erythroblasts and megaloblasts (Figure 4C). In addition, although the o-dianisidine staining of WT erythroblasts was normal, MT erythroblasts were only slightly stained (Figure 4D), suggesting that heme synthesis was highly impaired in MT erythroblasts. Furthermore, qRT-PCR analysis with the primers shown in supplemental Table 2 demonstrated that HBB, HBG, HO-1, and ALAS2 expression in MT erythroblasts was significantly decreased (Figure 4E; supplemental Figure 4G). By contrast, the expression of these genes in WT erythroblasts was comparable to that in the control erythroblasts.

Impaired erythropoiesis in MT iPSCs. (A) Colony formation assay on day 14 of hematopoietic differentiation. Each line was tested in 3 independent experiments. (B) Erythroid and mixed colony counts in panel A. (C) Representative images of erythroid (left) and mixed (right) colonies derived from WT1-iPSC2. Magnification of the objective lens: ×4. Bars represent 200 μm. (D) Erythroid and mixed colony counts in MT HPCs transduced with WT ALAS2. Each line was tested in 3 independent experiments. (E) Percentages of CD43+ hematopoietic cells. Each line was tested in 3 independent experiments. (F) Percentages of CD235a+ erythroblasts. Each line was tested in 3 independent experiments. All data are presented as the mean ± standard error of the mean. P-values were calculated by using the unpaired, 2-tailed Student t test. ****P < .0001; N.S., not significant.
Impaired erythropoiesis in MT iPSCs. (A) Colony formation assay on day 14 of hematopoietic differentiation. Each line was tested in 3 independent experiments. (B) Erythroid and mixed colony counts in panel A. (C) Representative images of erythroid (left) and mixed (right) colonies derived from WT1-iPSC2. Magnification of the objective lens: ×4. Bars represent 200 μm. (D) Erythroid and mixed colony counts in MT HPCs transduced with WT ALAS2. Each line was tested in 3 independent experiments. (E) Percentages of CD43+ hematopoietic cells. Each line was tested in 3 independent experiments. (F) Percentages of CD235a+ erythroblasts. Each line was tested in 3 independent experiments. All data are presented as the mean ± standard error of the mean. P-values were calculated by using the unpaired, 2-tailed Student t test. ****P < .0001; N.S., not significant.
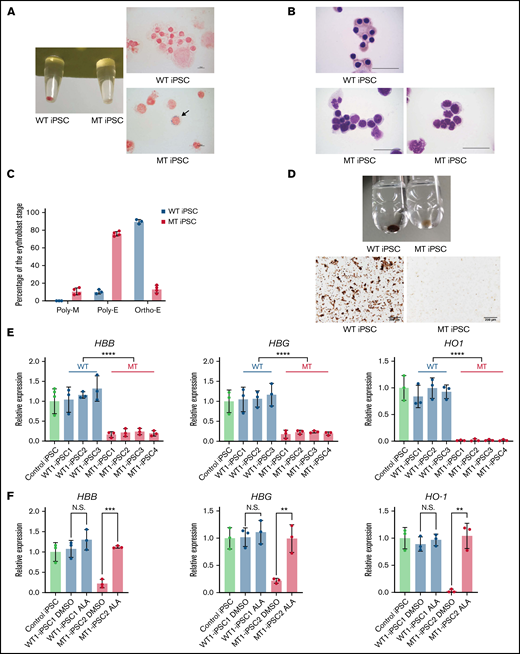
Maturation defects of erythroblasts derived from MT iPSCs. (A) The pellets (left) and iron staining (right) of erythroblasts derived from WT1-iPSC2 and MT1-iPSC3. The arrow indicates a ring sideroblast. CD235a+ cells were sorted on day 34 by FACS. Magnification of the objective lens: ×100. Bars represent 10 μm. (B) May Grunwald-Giemsa staining of erythroblasts derived from WT1-iPSC2 (top) and MT1-iPSC4 (bottom). CD235a+ cells were sorted on day 34 by FACS. Magnification of the objective lens: ×100. Bars represent 25 μm. (C) Percentages of polychromatic megaloblasts (Poly-M), polychromatic erythroblasts (Poly-E), and orthochromatic erythroblasts (Ortho-E) from 3 WT and 4 MT iPSC lines. (D) The pellets (top) and photomicrographs (bottom) of o-dianisidine–stained erythroblasts derived from WT1-iPSC3 and MT1-iPSC3 (bottom). CD235a+ cells were sorted on day 34 by FACS. Magnification of the objective lens: ×10. Bars represent, 200 μm. (E) The expression levels of HBB, HBG, and HO1 genes in erythroblasts derived from 1 control iPSC line, 3 WT iPSC lines, and 4 MT iPSC lines. Each line was tested in 3 independent experiments. Expression levels were normalized to the level of GAPDH. (F) The expression levels of HBB, HBG, and HO1 genes in WT1-iPSC1– and MT1–iPSC2-derived erythroblasts treated with DMSO or ALA relative to the expression levels of nontreated erythroblasts derived from a control iPSC line. Each line was tested in 3 independent experiments. Expression levels were normalized to the level of GAPDH. All data are presented as the mean ± standard error of the mean. P-values were calculated by using the unpaired, 2-tailed Student t test. **P < .01; ***P < .001; ****P < .0001; N.S., not significant. DMSO, dimethyl sulfoxide.
Maturation defects of erythroblasts derived from MT iPSCs. (A) The pellets (left) and iron staining (right) of erythroblasts derived from WT1-iPSC2 and MT1-iPSC3. The arrow indicates a ring sideroblast. CD235a+ cells were sorted on day 34 by FACS. Magnification of the objective lens: ×100. Bars represent 10 μm. (B) May Grunwald-Giemsa staining of erythroblasts derived from WT1-iPSC2 (top) and MT1-iPSC4 (bottom). CD235a+ cells were sorted on day 34 by FACS. Magnification of the objective lens: ×100. Bars represent 25 μm. (C) Percentages of polychromatic megaloblasts (Poly-M), polychromatic erythroblasts (Poly-E), and orthochromatic erythroblasts (Ortho-E) from 3 WT and 4 MT iPSC lines. (D) The pellets (top) and photomicrographs (bottom) of o-dianisidine–stained erythroblasts derived from WT1-iPSC3 and MT1-iPSC3 (bottom). CD235a+ cells were sorted on day 34 by FACS. Magnification of the objective lens: ×10. Bars represent, 200 μm. (E) The expression levels of HBB, HBG, and HO1 genes in erythroblasts derived from 1 control iPSC line, 3 WT iPSC lines, and 4 MT iPSC lines. Each line was tested in 3 independent experiments. Expression levels were normalized to the level of GAPDH. (F) The expression levels of HBB, HBG, and HO1 genes in WT1-iPSC1– and MT1–iPSC2-derived erythroblasts treated with DMSO or ALA relative to the expression levels of nontreated erythroblasts derived from a control iPSC line. Each line was tested in 3 independent experiments. Expression levels were normalized to the level of GAPDH. All data are presented as the mean ± standard error of the mean. P-values were calculated by using the unpaired, 2-tailed Student t test. **P < .01; ***P < .001; ****P < .0001; N.S., not significant. DMSO, dimethyl sulfoxide.
ALA improves erythroid differentiation capacity
We next explored the potential application of our iPSC-based disease model for drug testing. Heme synthesis starts with the polymerization of succinyl-CoA and glycine, followed by the synthesis of ALA in the mitochondria. ALAS2 encodes an enzyme that catalyzes the first step in the heme synthetic pathway. Thus, the administration of ALA to MT HPCs is expected to improve the capacity for heme synthesis. Indeed, we observed a marked increase in the erythroid differentiation of MT iPSCs after the administration of ALA from the beginning of erythroid differentiation (supplemental Figure 5A-C). In addition, the pellet of these erythroid cells was red, supporting the conclusion that ALA restored heme biosynthesis (supplemental Figure 5D). Moreover, qRT-PCR analysis demonstrated that ALA administration restored the expression of HBB, HBG, and HO1 (Figure 4F). These results support the use of our iPSC model for drug testing.
Expression of erythroid maturation-related genes is impaired in MT erythroblasts
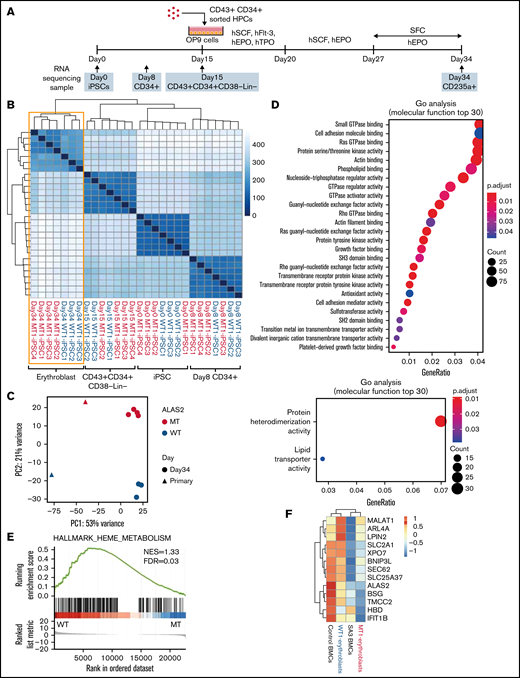
To compare the transcriptional expression patterns during erythroid differentiation in WT and MT cells, we performed RNA-seq of the iPSC, CD34+ cells on day 8; CD43+CD34+CD38−Lin− cells on day 15; and CD235a+ erythroblasts on day 34 in the 3 WT and 4 MT iPSC lines (Figure 5A). No differences were observed between the WT and MT cells at the stages of iPSCs, CD34+ cells, and CD43+CD34+38−Lin− cells. However, erythroblasts were divided into 2 clusters according to hierarchical clustering (Figure 5B). A principal component analysis of differentiated erythroblasts from the 7 iPSC lines and bone marrow erythroblasts from a healthy donor and SA3 revealed that iPSC-derived erythroblasts had cluster patterns similar to corresponding primary bone marrow erythroblasts. MT erythroblasts, and SA3 bone marrow erythroblasts were located on the positive side of PC2, whereas WT erythroblasts and healthy donor bone marrow erythroblasts were located on the negative side (Figure 5C). To verify this observation, the cells were assessed by using Gene Ontology (GO) analysis of the RNA-seq data. The upregulated genes in WT cells showed enrichment for mature erythroblast-related GO terms including Ras GTPase binding, ρ GTPase binding, and actin binding, and iron-transporter–regulated GO terms, such as transition metal ion transmembrane transporter activity (Figure 5D). This analysis indicated that WT erythroblasts differentiated into more mature stages than MT erythroblasts, despite the use of the same differentiation method. We also determined the enrichment of gene expression in WT erythroblasts and MT erythroblasts using gene set enrichment analysis for a set of genes differentially expressed during erythroid differentiation. The heme metabolism data set was significantly enriched in WT erythroblasts (Figure 5E; supplemental Table 3). Hierarchical clustering analysis of iPSC-derived and bone marrow erythroblasts for genes specifically expressed in early orthochromatic erythroblasts was performed (Figure 5F).28 The expression of these genes was lower in MT erythroblasts and SA3 bone marrow erythroblasts than that in WT erythroblasts and healthy donor bone marrow erythroblasts. These gene expression data support the findings of the phenotype experiments, which indicated that erythroblasts derived from MT iPSCs stopped at the immature erythroid stage.
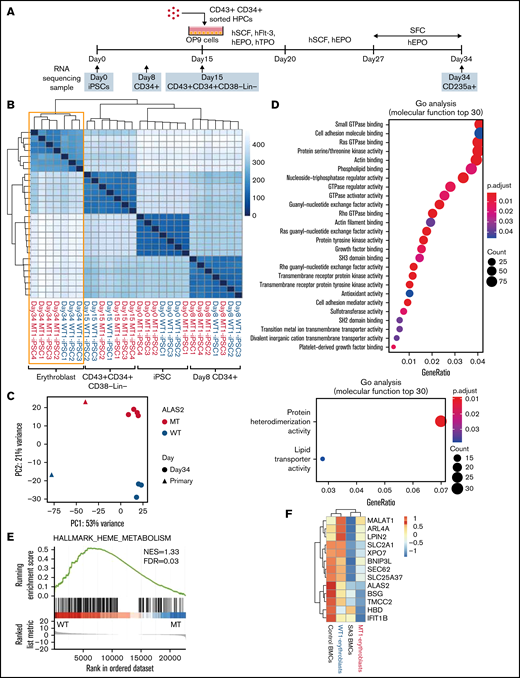
Gene expression in erythroblasts derived from WT and MT iPSCs. (A) Schema of the protocol for the erythroid differentiation from iPSCs and sample collection time points. Images of HPCs and OP9 cells were created using BioRender.com. (B) A heat map of the sample-to-sample distance and hierarchical clustering of iPSCs, CD34+ cells, CD43+CD34+CD38−Lin− cells, and erythroblasts derived from 3 WT and 4 MT iPSC lines with RNA-seq. (C) Principal component analysis of erythroblasts derived from the 3 WT and 4 MT iPSC lines and bone marrow erythroblasts from a healthy donor and SA3. (D) Two GO analyses showing molecular function terms enriched for WT erythroblasts (top) and MT erythroblasts (bottom). Differentially expressed genes between WT erythroblasts and MT erythroblasts were used. (|fold change| >2; adjusted P < .05). (E) Gene set enrichment analysis of the heme metabolism data set showed an enrichment of WT erythroblasts. (F) A heat map of 13 genes characteristically expressed at the orthochromatic stage in the erythroblasts described in panel C. For WT1-erythroblasts and MT1-erythroblasts, the heat map shows the average expression levels of the 3 and 4 lines, respectively.
Gene expression in erythroblasts derived from WT and MT iPSCs. (A) Schema of the protocol for the erythroid differentiation from iPSCs and sample collection time points. Images of HPCs and OP9 cells were created using BioRender.com. (B) A heat map of the sample-to-sample distance and hierarchical clustering of iPSCs, CD34+ cells, CD43+CD34+CD38−Lin− cells, and erythroblasts derived from 3 WT and 4 MT iPSC lines with RNA-seq. (C) Principal component analysis of erythroblasts derived from the 3 WT and 4 MT iPSC lines and bone marrow erythroblasts from a healthy donor and SA3. (D) Two GO analyses showing molecular function terms enriched for WT erythroblasts (top) and MT erythroblasts (bottom). Differentially expressed genes between WT erythroblasts and MT erythroblasts were used. (|fold change| >2; adjusted P < .05). (E) Gene set enrichment analysis of the heme metabolism data set showed an enrichment of WT erythroblasts. (F) A heat map of 13 genes characteristically expressed at the orthochromatic stage in the erythroblasts described in panel C. For WT1-erythroblasts and MT1-erythroblasts, the heat map shows the average expression levels of the 3 and 4 lines, respectively.
AZA treatment ameliorates heme synthesis failure and erythroid differentiation defects in MT iPSCs
We hypothesized that the pharmacological reactivation of the silent WT inactive X chromosome in the ALAS2 gene could ameliorate heme synthesis failure in patients with XLSA. In a previous study, it was suggested that the administration of a DNA demethylating agent, decitabine, reactivates the silent LAMP2 gene in iPSCs in female patients with Danon disease.29 Notably, in that study, other silent WT X-chromosome genes, including ALAS2, were also demethylated by decitabine. Therefore, we administered decitabine to the erythroid differentiation culture medium but found no improvement in the erythroid differentiation capacity of MT HPCs (data not shown). Next, we administered another DNA demethylating agent, AZA, at the erythroid differentiation stage (Figure 6A). The administration of AZA significantly improved the generation of CD235a+ erythroblasts in MT HPCs (Figure 6B; supplemental Figure 5E). Moreover, with o-dianisidine staining, erythroblasts differentiated from MT HPCs without AZA showed no positivity, whereas a considerable number of the same cells with AZA were positively stained (Figure 6C). We collected mRNA from AZA-treated and dimethyl sulfoxide control erythroblasts and performed complementary DNA sequencing of the ALAS2 gene. AZA reactivated the silent WT ALAS2 allele in MT HPCs and the silent mutant ALAS2 allele in active WT HPCs, suggesting the demethylation of the ALAS2 gene after AZA treatment (Figure 6D; supplemental Figure 5F). We also performed CFU assays with AZA treatment using iPSC-derived HPCs. There was no difference in the total colony counts between treated and control cells (Figure 6E). No erythroid colony and few mixed colonies were observed in MT HPCs with dimethyl sulfoxide. In contrast, erythroid and mixed colony formation was significantly improved by AZA treatment in MT HPCs, although it did not reach the levels seen in WT HPCs (Figure 6F-G). These results show that AZA treatment ameliorates the defective heme synthesis and ineffective erythropoiesis in MT HPCs.

Amelioration of heme synthesis failure and maturation defects of MT erythroblasts with AZA treatment. (A) Schema of a protocol for erythroid differentiation with AZA administration. The image of sorted HPCs was created using BioRender.com. (B) Percentages of CD235a+ erythroblasts treated with DMSO or AZA. The generation of CD235a+ erythroblasts in MT HPCs was significantly improved after the administration of AZA, whereas the generation of CD235a+ erythroblasts in WT HPCs was unchanged. Each line was tested in 3 independent experiments. (C) The pellets of unstained (top left) and o-dianisidine–stained (top right) erythroblasts derived from MT1-iPSC2 treated with DMSO or AZA. o-Dianisidine–stained images of erythroblasts derived from WT1-iPSC1 (left) and MT1-iPSC2 (right) treated with DMSO or AZA. CD235a+ cells were sorted on day 34 using FACS. Magnification of the objective lens: ×20. Bars represent 100 μm. (D) Representative Sanger sequencing data of ALAS2 complementary DNA from erythroblasts derived from WT iPSC (WT1-iPSC1) and MT iPSC (MT1-iPSC2) lines treated with DMSO or AZA. (E) Colony formation assay on day 15 of hematopoietic differentiation in WT1-iPSC1– and MT1-iPSC2–derived HPCs treated with DMSO or AZA. Each line was tested in 3 independent experiments. AZA 100, 100 nM AZA; AZA 500, 500 nM AZA. (F) Erythroid and mixed colony counts in panel E. (G) A representative image of mixed colonies derived from MT1-iPSC2 with AZA. Magnification of the objective lens: ×4. Bars represent 200 μm. All data are presented as the mean ± standard error of the mean. P-values were calculated using 1-way analysis of variance with Tukey’s correction and an unpaired, 2-tailed Student t test. *P < .05; **P < .01; N.S., not significant. DMSO, dimethyl sulfoxide.
Amelioration of heme synthesis failure and maturation defects of MT erythroblasts with AZA treatment. (A) Schema of a protocol for erythroid differentiation with AZA administration. The image of sorted HPCs was created using BioRender.com. (B) Percentages of CD235a+ erythroblasts treated with DMSO or AZA. The generation of CD235a+ erythroblasts in MT HPCs was significantly improved after the administration of AZA, whereas the generation of CD235a+ erythroblasts in WT HPCs was unchanged. Each line was tested in 3 independent experiments. (C) The pellets of unstained (top left) and o-dianisidine–stained (top right) erythroblasts derived from MT1-iPSC2 treated with DMSO or AZA. o-Dianisidine–stained images of erythroblasts derived from WT1-iPSC1 (left) and MT1-iPSC2 (right) treated with DMSO or AZA. CD235a+ cells were sorted on day 34 using FACS. Magnification of the objective lens: ×20. Bars represent 100 μm. (D) Representative Sanger sequencing data of ALAS2 complementary DNA from erythroblasts derived from WT iPSC (WT1-iPSC1) and MT iPSC (MT1-iPSC2) lines treated with DMSO or AZA. (E) Colony formation assay on day 15 of hematopoietic differentiation in WT1-iPSC1– and MT1-iPSC2–derived HPCs treated with DMSO or AZA. Each line was tested in 3 independent experiments. AZA 100, 100 nM AZA; AZA 500, 500 nM AZA. (F) Erythroid and mixed colony counts in panel E. (G) A representative image of mixed colonies derived from MT1-iPSC2 with AZA. Magnification of the objective lens: ×4. Bars represent 200 μm. All data are presented as the mean ± standard error of the mean. P-values were calculated using 1-way analysis of variance with Tukey’s correction and an unpaired, 2-tailed Student t test. *P < .05; **P < .01; N.S., not significant. DMSO, dimethyl sulfoxide.
Discussion
Most female patients with XLSA exhibit a severe anemia phenotype, as observed in the patients presented in this study.5,30,31 This pathology is caused by an increase in erythroblasts expressing a markedly defective mutant ALAS2 enzyme in the bone marrow because of the skewed inactivation of the X chromosome harboring the WT ALAS2 gene in hematopoietic stem cells later in life. In such cases, few mutant erythroblasts in which mutant ALAS2 is expressed differentiate into reticulocytes and reach circulation. Therefore, peripheral blood erythrocytes mainly represent descendant cells of the small number of clones with an active WT ALAS2 allele.5,32 Consistent with these reports, both CD34+ and CD235a+ bone marrow cells from SA2 and SA3 showed preferential inactivation of the X chromosome carrying WT ALAS2. By contrast, peripheral blood erythrocytes from SA2 and SA3 were derived exclusively from a few erythroblasts expressing the WT ALAS2 gene.
WT and MT iPSC lines have been established for several X-linked diseases in which X-chromosome inactivation is maintained after reprogramming.29,33,34 However, it remains unclear whether the same is true for XLSA. In the present study, we generated iPSC lines with an active mutant ALAS2 allele and isogenic iPSC lines with an active WT ALAS2 allele from the peripheral blood cells of 3 female patients with XLSA. All female XLSA-iPSC lines were found to have extremely skewed X-chromosome inactivation patterns and consistently expressed either the WT or mutant allele, but not both, of the ALAS2 gene after erythroid differentiation.
Consistent with the severe ineffective erythropoiesis in our patients with XLSA, MT HPCs were difficult to differentiate into erythroblasts in our feeder-free condition, because most of them died at an early stage of the erythroid differentiation. In a previous report, erythroblasts derived from XLSA-iPSCs exhibited ring sideroblasts by coculturing with stromal cells.16 Therefore, we adopted the OP9 coculture method to perform precise morphological and gene expression analyses of the MT erythroblasts. According to another report, the coculture of adult CD34+ cells with OP9 feeder cells increases the proliferation ability of erythroid cells by delaying erythroid differentiation.35 We therefore speculated that stromal cell coculture maintains MT erythroblasts in a more immature state than the feeder-free condition, preventing their death as maturation proceeds. MT erythroblasts failed to synthesize heme and exhibited maturation defects with ring sideroblast formation in contrast to WT erythroblasts (Figure 7). RNA-seq revealed that MT erythroblasts showed decreased expression of genes associated with erythroid maturation, which is consistent with the immature morphology of MT erythroblasts and female patient bone marrow erythroblasts. To the best of our knowledge, this is the first report to demonstrate the defective erythroid differentiation of ALAS2-affected HPCs from female patients with XLSA with a heterozygous ALAS2 missense mutation. A previous report of ALAS2-knockout murine embryonic stem cells showed that ALAS2 deficiency does not interfere with the maturation of definitive erythroid cells.15 One explanation for this phenomenon could be the differences in mice and human patients with XLSA. In a previous study, iPSCs derived from male patients with XLSA differentiated into orthochromatic erythroblasts, and the erythroid maturation defect was not observed.16 Another study showed that human iPSC-derived erythroid progenitor cells and XLSA bone marrow cells lacking the ALAS2 enhancer region had increased expression levels of antiapoptosis genes.18 However, our MT erythroblasts stopped at the early stage of erythroid differentiation and did not show the same expression profiles in RNA-seq (data not shown). Although the reason for the observed discrepancies is unclear, it may be because of the degree of ALAS2 functional loss between our model and the previous models. Alternatively, sex differences could be a factor. Overall, our iPSC model may be a useful tool for understanding the roles of ALAS2 and heme synthesis in maintaining the erythroid cell transcriptome and morphology associated with XLSA.

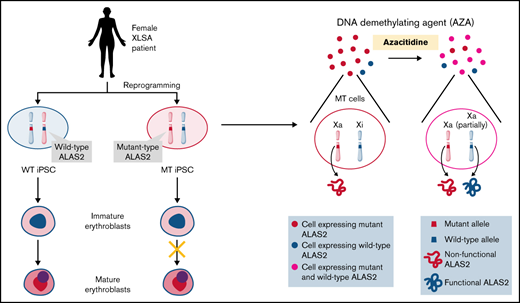
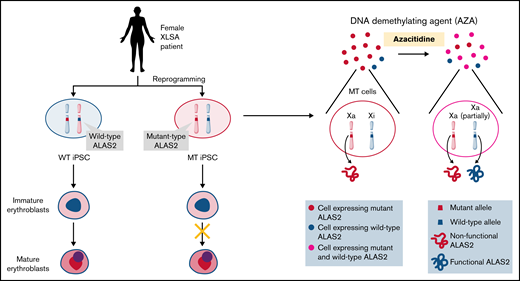
Schematic representation of our results. We established 2 types of iPSC lines derived from individual female patients with XLSA harboring the ALAS2-R227C mutation. WT iPSCs showed mature erythroid differentiation, and MT iPSCs stopped at the immature erythroblast stage, recapitulating the pathogenesis of XLSA. AZA administration reactivated the silent WT ALAS2 allele in MT HPCs and ameliorated erythroid differentiation defects. The figure was created using BioRender.com.
Schematic representation of our results. We established 2 types of iPSC lines derived from individual female patients with XLSA harboring the ALAS2-R227C mutation. WT iPSCs showed mature erythroid differentiation, and MT iPSCs stopped at the immature erythroblast stage, recapitulating the pathogenesis of XLSA. AZA administration reactivated the silent WT ALAS2 allele in MT HPCs and ameliorated erythroid differentiation defects. The figure was created using BioRender.com.
The responsiveness of patients with XLSA to pyridoxine supplementation varies considerably according to the nature and position of the amino acid substitution in ALAS2.11 In our model, the supplementation of pharmacologic levels of pyridoxal hydrochloride or pyridoxine hydrochloride did not improve the erythroid differentiation capacity of MT HPCs, coinciding with the unresponsiveness to pyridoxine in this patient (data not shown). This finding suggests that our iPSC-based platform is useful for precisely assessing pyridoxine sensitivity in patients with XLSA with various ALAS2 mutations.
Previous reports have shown that ALA restores heme synthesis by human iPSC-derived erythroid progenitor cells and K562 cells.36 Using our iPSC platform, we demonstrated that ALA not only restores heme synthesis but also ameliorates the erythroid differentiation efficiency of MT iPSC lines in vitro. However, the oral administration of ALA did not improve anemia in our patients, consistent with a reported case.12 This may be because the concentration of ALA in the bone marrow was not sufficient to improve the pathophysiology of MT cells. Drugs that increase the cellular uptake of ALA, especially in early erythroid cells, may improve the drug efficacy. In addition, a dose-escalation clinical study may be useful in assessing the safety and efficacy of ALA in pyridoxine-resistant patients with XLSA.
Last, we focused on the unusual disease mechanism of XLSA in females, which is caused by the acquired skewing of X-chromosome inactivation in hematopoietic stem cells. We speculated that the restoration of the expression of WT ALAS2 in a considerable number of affected erythroblasts could ameliorate disease phenotypes in female patients with XLSA. This study provides the first proof of concept that the silent WT ALAS2 allele in female subjects could be partially reactivated by the DNA demethylating agent AZA, which serves as a potential novel therapeutic approach for the treatment of refractory anemia in female patients with XLSA (Figure 7). Although the demethylating effect of AZA is global and off-target side effects may occur in both hematopoietic and nonhematopoietic tissues, AZA is currently being used in clinical practice to treat hematological malignancies and has shown acceptable adverse effects. However, the efficacy of AZA in our XLSA model is limited, and further studies using DNA demethylating agents are needed for future therapeutic use.
In summary, this study provides novel biological and therapeutic insights into XLSA. Using a patient-specific iPSC platform, we showed that a severe loss-of-function ALAS2 mutation could cause maturation defects in erythroid cells that resemble the phenotype of female XLSA. Furthermore, we revealed that the demethylating agent AZA ameliorates the disease phenotype of female XLSA by reactivating the silenced WT ALAS2 allele. Considering the acceptable toxicity in some patients with cancer, DNA demethylating agents may promise new therapeutic candidates for female patients with XLSA.
Acknowledgments
The authors thank Peter Karagiannis for proofreading the manuscript, Kaoru Shimizu and Mikako Marx-Mori for their administrative support, and Editage (www.editage.com) for editing the English.
This work was supported in part by grants from the Research Center Network for Realization of Regenerative Medicine (JP19bm0104001) [Y.Y.], JP19bm0804004 [Y.Y., K.C.], JP19bm0804008 [Y.Y.], and JP20bm0804022 [Y.Y.]) and the Project for Cancer Research and Therapeutic Evolution (JP19cm0106235 [Y.Y., K.C.]), provided by the Japan Agency for Medical Research and Development; grants from the Mochida Memorial Foundation for Medical and Pharmaceutical Research (Y.Y., K.C.), the Yasuda Memorial Medical Foundation (Y.Y.), the Kobayashi Foundation for Cancer Research (Y.Y.), the Daiichi Sankyo Foundation of Life Science (Y.Y.), the Leducq Foundation (18CVD05) (Y.Y.), and the iPS Cell Research Fund (Y.Y.). Y.M. is a research fellow of the Japan Society for the Promotion of Science (17J07616).
Authorship
Contributions: Y.M., K.C., and Y.Y. conceived and designed the research; Y.M. performed most of the experiments, analyzed the data, and wrote the manuscript; K.C. performed some experiments, analyzed the data, and wrote the manuscript; C.O. analyzed the RNA-seq data; M. Nishikawa, M. Narita, and A.I. provided essential experimental support; M.Y-M., K.N., and M. Nagao assisted with the hematopoietic cell staining; H.K. collected patient blood samples and advised on the experimental design; A.T-K. advised on the experimental design and wrote the manuscript; Y.Y. supervised the overall research and wrote the manuscript; and all authors interpreted the data and reviewed and approved the final manuscript.
Conflict-of-interest disclosure: Y.M., K.C., H.K., A.T-K., and Y.Y. are the inventors of JP patent applications (JP2019/525414 and JP2020/218557). H.K. and A.T-K. received honoraria from Nippon Shinyaku outside the submitted work. A.T-K. and Y.Y. received grants from Nippon Shinyaku outside the submitted work. C.O., M.Y-M., M. Nishikawa, M. Narita, K.N., and M. Nagao declare no competing financial interests.
Correspondence: Kazuhisa Chonabayashi, Department of Hematology and Oncology, Graduate School of Medicine, Kyoto University, 54 Kawahara-cho, Shogoin, Sakyo-ku, Kyoto 606-8507, Japan; e-mail: kchona@kuhp.kyoto-u.ac.jp; and Yoshinori Yoshida, Department of Cell Growth and Differentiation, Center for iPS Cell Research and Application, Kyoto University, 53 Kawahara-cho, Shogoin, Sakyo-ku, Kyoto 606-8507, Japan; e-mail: yoshinor@cira.kyoto-u.ac.jp