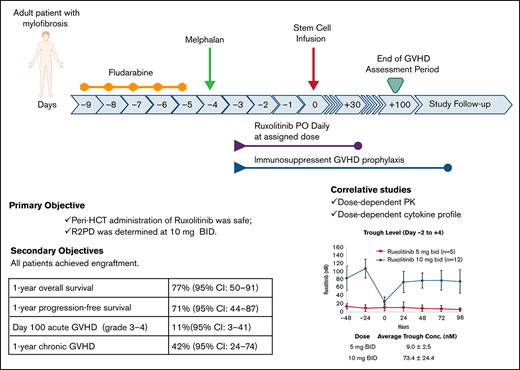
Key Points
Peritransplantation ruxolitinib was safe and well tolerated in patients with myelofibrosis, with MTD determined as 10 mg twice daily.

Abstract
We report results of our prospective pilot trial evaluating safety/feasibility of peritransplantation ruxolitinib for myelofibrosis treatment. Primary objectives were to determine safety and maximum tolerated dose (MTD) of ruxolitinib. Ruxolitinib was administered at 2 dose levels (DLs) of 5 and 10 mg twice daily, with fludarabine/melphalan conditioning regimen and tacrolimus/sirolimus graft-versus-host disease (GVHD) prophylaxis. We enrolled 6 and 12 patients at DL1 and DL2, respectively. Median age at transplantation was 65 years (range, 25-73). Per Dynamic International Prognostic Scoring System, 4 patients were high and 14 intermediate risk. Peripheral blood stem cells were graft source from matched sibling (n = 5) or unrelated (n = 13) donor. At each DL, 1 patient developed dose-limiting toxicities (DLTs): grade 3 cardiac and gastrointestinal with grade 4 pulmonary DLTs in DL1, and grade 3 kidney injury in DL2. All patients achieved engraftment. Grade 2 to 4 and 3 to 4 acute GVHD cumulative incidence was 17% (95% confidence interval [CI], 6-47) and 11% (95% CI, 3-41), respectively. Cumulative incidence of 1-year chronic GVHD was 42% (95% CI, 24-74). With 22.6-month (range, 6.2-25.8) median follow-up in surviving patients, 1-year overall and progression-free survival were 77% (95% CI, 50-91) and 71% (95% CI, 44-87), respectively. Causes of death (n = 4) were cardiac arrest, GVHD, respiratory failure, and refractory GVHD of liver. Our results show peritransplantation ruxolitinib is safe and well tolerated at MTD of 10 mg twice daily and associated with dose-dependent pharmacokinetic and cytokine profile. Early efficacy data are highly promising in high-risk older patients with myelofibrosis. This trial was registered at www.clinicaltrials.gov as #NCT02917096.
Introduction
Myelofibrosis (MF) is a clonal myeloproliferative neoplasm that can be classified as primary or secondary to either essential thrombocythemia or polycythemia vera, characterized by varying degrees of cytopenia, bone marrow (BM) fibrosis, and heterogenous symptom burden/prognosis.1 Dysregulation of JAK-STAT pathway is the hallmark of MF and has become the therapeutic target for this disease.2,3 Ruxolitinib is a potent JAK1/2 inhibitor that was approved in the United States in 2011 for treatment of intermediate- or high-risk MF, based on results of 2 phase 3 trials: double-blind COMFORT-I and open-label COMFORT-II.4,5 Although JAK inhibitors are currently the best available therapy for splenomegaly and constitutional symptoms associated with MF, treatment with these drugs has no lasting disease-modifying effect. Allogeneic hematopoietic cell transplantation (HCT) remains the only potential curative therapy for MF and is increasingly offered for patients with intermediate- or high-risk disease.6,7 Unfortunately, HCT is associated with a significant risk of transplantation-related morbidity and mortality, with 1 of the most serious complications being graft-versus-host disease (GVHD).
Ruxolitinib, through its inhibition of JAK1/2 signaling, was shown to reduce GVHD in mice by inducing durable and profound specific T-cell tolerance and preserving regulatory T cells (Tregs).8,9 Results of a small pilot study treating steroid-refractory (SR) severe acute and chronic GVHD with ruxolitinib showed excellent clinical activity accompanied by reduction in serum proinflammatory cytokines and increase in Foxp3+ Tregs.9 Multiple prospective and retrospective studies have confirmed the safety and clinical efficacy of ruxolitinib for treatment of SR GVHD. Specifically, REACH1 and REACH2 trials led to approval of ruxolitinib for treatment of SR acute GVHD.10
At present, the optimal use of ruxolitinib in patients with MF immediately before and after HCT is unknown. Pre-HCT administration of ruxolitinib has been shown to have beneficial effects on spleen size and potentially engraftment.11-14 However, ruxolitinib is generally tapered off before start of conditioning, because sudden stoppage of the drug is often associated with a cytokine storm-like condition called ruxolitinib withdrawal syndrome.15 Results of a pilot study (N = 12) by Kroger et al16 showed continuing ruxolitinib at 5 mg twice daily through peri-HCT period until stable engraftment is feasible, with a lower incidence of GVHD, when cyclosporine, mycophenolate mofetil, and anti–T-cell globulin were administered as GVHD prophylaxis. Outcomes of another prospective study in which ruxolitinib was added to posttransplantation cyclophosphamide from day 7 to +100 in 20 patients with MF showed acceptable acute and chronic GVHD rates; however, ruxolitinib administration was associated with primary graft failure (n = 1), death before engraftment (n = 2), and severe poor graft function (n = 11).17
At City of Hope (COH), we pioneered reduced-intensity conditioning (RIC) HCT using fludarabine and melphalan (Flu/Mel) as conditioning regimen combined with tacrolimus and sirolimus (Tac/Sir) as GVHD prophylaxis, with excellent survival.18-20 We previously reported on the clinical outcome of a large cohort (n = 110) of patients with MF who underwent HCT with this regimen at our center and showed excellent 5-year overall survival (OS) of 64%, with low risk of relapse (17%).21 Flu/Mel has been shown to be highly effective in myeloid malignancies, and Tac/Sir is associated with faster engraftment compared with commonly used Tac/methotrexate.22 Therefore, Tac/Sir-based GVHD prophylaxis may limit potential myelosuppression when combined with the JAK1/2 inhibitor ruxolitinib.
Based on the hypothesis that continuing ruxolitinib during HCT may prevent ruxolitinib withdrawal syndrome, improve engraftment, and reduce GVHD, we conducted a pilot phase 1 trial evaluating the safety and efficacy of peri-HCT administration of ruxolitinib in patients with MF who were eligible for RIC HCT.
Methods
Protocol
This prospective single-center open-label clinical trial was approved by the COH Institutional Review Board. An assurance was filed with and approved by the US Department of Health and Human Services. Informed consent was obtained for all study participants in compliance with the Declaration of Helsinki.
Study design
In this pilot study with an expansion cohort, adult patients with MF received peri-HCT ruxolitinib administration (day −3 to +30) along with our standard RIC of Flu/Mel and Tac/Sir as GVHD prophylaxis. The primary objectives were to determine the maximum tolerated dose (MTD) and recommended phase 2 dose (RP2D) of ruxolitinib and determine its safety. Secondary objectives were grade 2 to 4 acute GVHD (cumulative incidence), chronic GVHD, donor cell engraftment (recovery of granulopoiesis and megakaryopoiesis), grade 3 to 4 infection, OS, progression-free survival (PFS), cumulative incidence of disease relapse or progression (cumulative incidence), and nonrelapse mortality.
We employed a rolling 6-dose escalation design, with rules similar to a standard 3 + 3 phase 1 design but allowing up to 6 patients to be treated at a dose level, with only 3 at risk for dose-limiting toxicities (DLTs) at any 1 time.23 Two dose levels (DLs) of 5 and 10 mg twice per day (DL1 and DL2, respectively) were examined. DLT was defined as any regimen-related grade 3 or 4 toxicity per Bearman criteria,24 any grade 4 neutropenia with fever or infection lasting >21 days, grade 4 neutropenia lasting >28 days (engraftment failure), any other regimen-related death, and any grade 5 sepsis-related toxicity that was assigned an attribution level of at least possibly related to ruxolitinib. MTD was based on the assessment of DLTs from the start of therapy to 45 days after hematopoietic cell infusion (15 days after last full dose of ruxolitinib).
Patients and treatment
Adult patients between 18 and 75 years of age with primary or secondary MF at intermediate-2– or high-risk per Dynamic International Prognostic Scoring System criteria25 who were scheduled to undergo their first allogeneic transplantation from an 8/8 HLA-matched (-A, -B, -C, or -DR by high resolution) donor were eligible for the study. Prior use of ruxolitinib was allowed.
As shown in Figure 1A, conditioning regimen consisted of fludarabine (25 mg/m2 per day, calculated per actual body weight) as a daily infusion from day −9 to −5. Mel was administered at a single dose of 140 mg/m2 (optional 100 mg/m2 for patients age >70 years) on day −4. Patients who were receiving ruxolitinib before conditioning continued it until day −10, but ruxolitinib administration at a lower dose during conditioning was allowed on a case-by-case basis. This decision was made out of abundance of caution and to avoid ruxolitinib withdrawal syndrome and potential interaction between ruxolitinib and conditioning chemotherapy. Ruxolitinib was then administered at the assigned DL after conditioning from day −3 to +30. Thereafter, ruxolitinib was tapered as follows: 5 mg daily for 5 days then stop for DL1, and 5 mg twice daily for 3 days then stop for DL2.
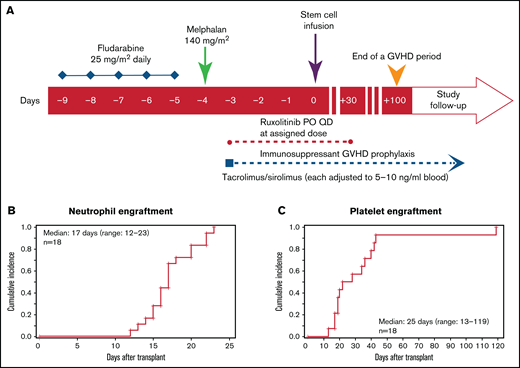
Conditioning regimen and engraftment. (A) Study schema. Peri-HCT ruxolitinib administration was from day −3 pre-HCT to day +30 post-HCT. Neutrophil (B) and platelet (C) engraftment post-HCT. aGVHD, acute GVHD; PO, orally; QD, once daily.
Conditioning regimen and engraftment. (A) Study schema. Peri-HCT ruxolitinib administration was from day −3 pre-HCT to day +30 post-HCT. Neutrophil (B) and platelet (C) engraftment post-HCT. aGVHD, acute GVHD; PO, orally; QD, once daily.
GVHD prophylaxis consisted of Tac (0.02 mg/kg/d continuous IV), beginning on day –3 and converting to oral dosing when the patient was able to tolerate and absorb oral medications. Sirolimus was administered at a 12 mg oral loading dose on day −3, followed by 4 mg orally as a single morning daily dose. Target serum level for both Tac and Sir is 5 to 10 ng/mL by high-performance liquid chromatography; levels were adjusted by treating physicians to maintain this range. In the absence of GVHD, the immunosuppressive taper was provided as per COH standard operating procedures.19,26 Infection monitoring practice included cytomegalovirus (CMV) polymerase chain reaction (twice per week starting on day +7 and continued until day +100) and hepatitis B/C and HIV tests at admission. Epstein-Barr virus and human herpesvirus 6 tests were performed if clinically indicated.
Statistical analysis
Patient demographic and baseline characteristics, including age, sex, medical history, and prior therapy, are summarized using descriptive statistics. For continuous variables, descriptive statistics (number, mean, standard deviation, standard error, median [range]) are provided. For categorical variables, patient counts and percentages are provided. Observed toxicities are summarized in terms of type (organ affected or laboratory determination), severity, time of onset, duration, probable association with study treatment, and reversibility or outcome. Cumulative incidence of acute and chronic GVHD was calculated using the Gray method, with prior death or relapse considered competing events. Survival estimates were calculated using the Kaplan-Meier method.
All cytokines and biomarkers were measured repeatedly over time. Median and range at each time point for the 2 DLs are displayed by box plot. Differences between the 2 DLs were examined by Wilcoxon rank sum test.
End point definition
Platelet engraftment was defined as the first of 7 consecutive days in which the platelet count was >10 × 109/L without transfusion support. Neutrophil engraftment was defined as absolute neutrophil count ≥0.5 × 103/μL achieved and sustained for 3 consecutive laboratory values on different days with no subsequent decline. GVHD-free/GVHD relapse–free survival was a post hoc end point defined as survival without grade 3 to 4 acute GVHD, moderate/severe chronic GVHD, relapse, progression, or death (resulting from any cause).27
Pharmacokinetics
Blood samples for pharmacokinetic studies were collected before morning ruxolitinib doses on days −2 through +5. Additional postdose samples were obtained after morning dose on day +5 at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, and 12 hours. Blood samples were kept on ice until plasma was separated by centrifugation at 1500× g (within 1 hour). Plasma samples were stored below −70°C until analysis. Plasma concentrations of ruxolitinib were measured using a liquid chromatography–tandem mass spectrometry assay. The analytical method was based on a previously reported assay by Veeraraghavan et al28 that has been validated over a concentration range of 0.2 to 250 ng/mL from a starting plasma volume of 50 μL. Ruxolitinib concentration vs time data were analyzed using standard noncompartmental analysis methods according to the rule of linear trapezoids. Noncompartmental pharmacokinetic parameters were derived for each patient and included trough concentrations, maximum plasma concentration, time to maximum plasma concentration, terminal-phase elimination half-life, area under the plasma concentration–time curve from 0 to t, and oral clearance. Individual parameters are summarized as means or medians, along with standard deviations or ranges.
Flow cytometry, plasma cytokines, and GVHD biomarkers
Peripheral blood samples were collected on days +21, +35, and +100 post-HCT. Peripheral blood mononuclear cells were isolated and cryopreserved following standard procedures. For Treg staining, peripheral blood mononuclear cells were surface stained for CD3, CD4, CD8, CD19, CD56, CD25, and CD127 (eBioscience) and intracellularly stained for Foxp3 (eBioscience) using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience). Flow cytometry was performed using the BD FACSCelesta (BD Biosciences), and data were analyzed using Flowjo software (TreeStar). Foxp3 values were based on isotype controls.
Serum samples, obtained on days +7, +21, +28, +42, and +100, were analyzed for 30 different cytokines using the Human Cytokine Thirty-Plex Antibody Magnetic Bead Kit (Invitrogen, Camarillo, CA) per manufacturer recommendations. The Flexmap 3D Luminex System (Luminex) was used for analysis, and cytokine concentrations were calculated using Bio-Plex Manager 6.2 software with a 5-parameter curve-fitting algorithm applied for standard curve calculations for duplicate samples.
Results
Patient and transplantation characteristics
A total of 18 patients were enrolled, of whom 12 (5 patients in DL1 and 7 in DL2) were receiving ruxolitinib treatment before conditioning regimen, and 6 were taken off ruxolitinib preconditioning. Ruxolitinib duration and continuation in patients who took ruxolitinib preconditioning are summarized in supplemental Table 1. Patient demographics and disease/transplant characteristics are summarized in Table 1. Briefly, median age was 65 years (range, 25-73) for all patients (53 years [range, 25-67] at DL1 [n = 6] and 69 years [range, 55-73] at DL2 [n = 12]). By Dynamic International Prognostic Scoring System criteria, 4 patients were high risk and the remaining 14 were intermediate-2 risk. By Mutation-Enhanced International Prognostic Scoring System 70, 14 patients were high risk, 1 was intermediate risk, and 3 did not have available data. By Mutation-Enhanced International Prognostic Scoring System 70 Plus, 7 patients were categorized as very high, 6 as high, and 2 as intermediate risk. Cytogenetic abnormalities were unfavorable in 5 and favorable in 13 patients. Driver mutations were JAK2 (n = 9), CALR (n = 4), and MPL (n = 1). High molecular risk mutations were present in 8 patients, 2 of whom carried 2 high molecular risk mutations. Time from diagnosis to HCT was 17.2 months (range, 3.0-74.5). All patients received mobilized peripheral blood stem cells from a matched related (n = 5) or unrelated (n = 13) donor at a median CD34+ cell dose of 6.0 × 106/kg (range, 3.9-9.1). Median follow-up duration for surviving patients was 22.6 months (range, 6.2-25.8). Only 1 patient had splenomegaly after HCT for poor counts. Ruxolitinib compliance through the intended treatment period of 34 days was 337.5 mg (range, 295-340) for DL1 and 680 mg (range, 500-740) for DL2.
Safety/toxicity
All patient completed treatment with ruxolitinib. Common adverse events (AEs), DLTs, and serious AEs are summarized in Table 2 according to DL. Of the first 3 patients in DL1, the first patient developed a DLT (severe mucositis resulting in airway obstruction with consequent respiratory failure and cardiac arrest resulting from pulseless electrical activity, from which he recovered, but he died of recurrent respiratory failure on day 48). After observing 1 DLT at DL1, we enrolled 5 additional patients in this arm, and once this dose level was deemed safe, we escalated the dose to 10 mg twice per day and enrolled 12 more patients at DL2. One patient in DL2 developed a DLT of acute kidney injury on day 23 requiring hemodialysis, which was thought to be due to thrombotic microangiopathy associated with Tac. The thrombotic microangiopathy was successfully treated with eculizumab. This patient completely recovered and became dialysis independent by day 103 of HCT. Overall majority of the AEs were grade 1 and 2. Serious AEs were reported in 8 patients (19 events), none of which was considered as being related to the study intervention.
Infectious complications from day −9 to +100 included CMV viremia (n = 3), respiratory infections (n = 3), and BK virus cystitis (n = 1). Three patients had bacteremia with gram-positive coccids, and 4 developed Clostridium difficile colitis.
Engraftment
All patients achieved neutrophil recovery at a median of 19 days (range, 13-23) at DL1 and 16 days (range, 12-22) at DL2 (Figure 1B). Platelet engraftment was achieved in 14 patients at a median of 25 days (range, 13-119; (20 days [range, 19-42] at DL1 and 28 days [range, 13-119] at DL2]; Figure 1C). Four patients did not achieve platelet transfusion independence; contributing factors were SR acute GVHD (n = 3) and CMV infection (n = 1). Engraftment analyses of evaluable patients (n = 16) demonstrated ≥95% donor chimerism for total BM cells, while BM T cells were all >90%, except for 1 patient at DL1 whose donor chimerism was 85% at day 30. At day 100, all patients had 99% donor chimerism for BM and BM T cells, except 1 patient in the DL1 arm with 95% chimerism. At 1 year, all patients had 100% donor chimerism either by blood or BM. None of the patients had secondary graft failure.
GVHD
Cumulative incidence of grade 2 to 4 and 3 to 4 acute GVHD at 100 days was 17% (95% confidence interval [CI], 6-47) and 11% (95% CI, 3-41), respectively, for the entire cohort. Number of cases and individual grades are summarized in Table 3. All 3 patients diagnosed with grade 2 to 4 acute GVHD developed GVHD before day +30 and during the ruxolitinib administration period. One patient with grade 2 to 4 GVHD underwent HCT from an HLA mismatched donor, and the other 2 patients could not receive Tac as GVHD prophylaxis because of kidney dysfunction. None of the patients in our cohort developed delayed-onset acute GVHD. The first-line treatment for acute GVHD was systemic corticosteroids. All patients with grade 2 to 4 GVHD (n = 3) developed SR acute GVHD requiring second-line agents basiliximab (n = 2) or extracorporeal photopheresis/infliximab/ruxolitinib (n = 1). Chronic GVHD developed in 50% of patients, with a median onset at 6.7 months (Table 3). Cumulative incidence of moderate/severe chronic GVHD at 1 year was 24% (95% CI, 10-55). Supplemental Table 2 summarizes organ involvement and severity of acute and chronic GVHD.
Survival outcomes
The 1-year OS and PFS rates for the entire cohort were 77% (95% CI, 50-91) and 71% (95% CI, 44-87), respectively. Cumulative incidence of relapse and nonrelapse mortality at 1 year was 6% (95% CI, 1-40) and 23% (95% CI, 10-54), respectively. Estimate of 1-year GVHD relapse–free survival was 52% (95% CI, 26-73). Causes of death were cardiac arrest (n = 1), GVHD (n = 2), and respiratory failure related to severe mucositis (n = 1).
Pharmacokinetics
As expected, the average trough levels of ruxolitinib from day −2 to +4 were higher at DL2 (9.0 ± 2.5 nM at DL1 and 73.4 ± 24.4 nM at DL2; P < .01; Figure 2A). On day 5, detailed pharmacokinetic samples were obtained at 11 different time points over 12 hours, which demonstrated maximum plasma concentration of 150.8 ± 39.9 nM at DL1 and 273.4 ± 78.4 nM at DL2 (P < .01) and area under the plasma concentration–time curve from 0 to ∞ of 546.2 ± 186.5 nM per hour at DL1 and 990.6 ± 373.5 nM per hour at DL2 (P < .01). Terminal-phase elimination half-life was similar between the 2 DLs: 2.3 ± 0.4 hours at DL1 and 2.2 ± 0.9 hours at DL2 (P = .74). Oral clearance was also similar between DL1 (32.8 ± 11.2 L per hour) and DL2 (36.4 ± 10.8 L per hour; P = .57; Figure 2B).

Ruxolitinib pharmacokinetics. Days −2 to +4 (A) and +5 (B). AUC, area under the plasma concentration–time curve; BID, twice daily; CL/F, oral clearance; Cmax, maximum plasma concentration; PK, pharmacokinetics; T1/2, terminal-phase elimination half-life.
Ruxolitinib pharmacokinetics. Days −2 to +4 (A) and +5 (B). AUC, area under the plasma concentration–time curve; BID, twice daily; CL/F, oral clearance; Cmax, maximum plasma concentration; PK, pharmacokinetics; T1/2, terminal-phase elimination half-life.
Immune reconstitution, plasma cytokines, and GVHD biomarkers
Recovery of total lymphocytes, CD4+ T cells, CD8+ T cells, NK cells, B cells, and Tregs is depicted in Figure 3. Median lymphocyte, CD4 T-cell, CD8 T-cell, NK cell (CD3−CD56+), B-cell (CD19+), and Treg (CD4+CD25+CD127−Foxp3+) counts (per μL) on day 100 for the entire cohort were 0.4 (range, 0.1-0.7), 106.6 (range, 15.8-323.6), 48.5 (range, 9.8-324.7), 103.9 (range, 52.4-163.3), 8.1 (range, 0.2-34.8), and 2.3 (range, 0.0-8.9), respectively. On day +21, we observed a trend (P = .07) toward faster recovery of CD3+ T cells at DL2 (median, 97.0; range, 29.2-714) compared with DL1 (median, 38.5; range, 21.6-62.7). The same trend was seen on recovery of CD8+ cells on day +21 (median, 22.8; range, 2.1-317.0 at DL2 vs median, 7.2; range, 3.1-12.5 at DL1; P = .06) and CD27+ memory B cells on day +35 (median, 0.7; range, 0.2-2.0 at DL2 vs median, 0.3; range, 0.2-0.5 at DL1; P = .09; Figure 3).

Comparison of immune reconstitution. Reconstitution of DL1 (5 mg twice daily) vs DL2 (10 mg twice daily) on days 21, 35, and 100 post-HCT for CD3+ T cells (A), CD3+ T cells (B), CD8+ T cells (C), Foxp3+ Tregs (D), CD27+ B cells (E), and CD56+ NK cells (F).
Comparison of immune reconstitution. Reconstitution of DL1 (5 mg twice daily) vs DL2 (10 mg twice daily) on days 21, 35, and 100 post-HCT for CD3+ T cells (A), CD3+ T cells (B), CD8+ T cells (C), Foxp3+ Tregs (D), CD27+ B cells (E), and CD56+ NK cells (F).
When plasma cytokine levels were compared between DL1 and DL2, we observed that interleukin-2 (IL-2), interferon-γ (IFN-γ), tumor necrosis factor α (TNF-α) were significantly lower at DL2 (unadjusted for multiple testing; P ≤ .05; supplemental Figure 1). No significant difference was detected in levels of GVHD biomarkers between DL1 and DL2 at the .05 level (supplemental Figure 2). Supplemental Figure 3 shows levels of GVHD biomarkers in the 3 patients with higher grades of acute GVHD compared with minimum, maximum, and median levels in patients at each dose level.
Discussion
We previously reported long-term outcomes of patients with MF who underwent HCT with Flu/Mel conditioning and Tac/Siro as GVHD prophylaxis with excellent outcomes and long-term remission.18-20 In the current study, we show in our high-risk cohort of patients with MF that addition of ruxolitinib to this regimen is safe and well tolerated and is associated with low rates of GVHD and promising survival. We identified the ruxolitinib dose of 10 mg twice per day as the RP2D, because the toxicities were acceptable, with majority being grade 1 toxicities. Incidence of infection was similar to what is expected in the HCT population. It is possible that our study design of limiting ruxolitinib to 30 days post-HCT might have contributed to the tolerability and lack of prolonged immunosuppression. Day +30 was selected to provide initial immune tolerance during the engraftment period. We chose not to continue ruxolitinib administration further than day +30 to avoid prolonged cytopenia and subsequent increased risk of infection. There was no immune flare after stopping ruxolitinib in our cohort, which could be attributable to the carefully planned tapering schedule and effective disease control within the first 30 days after HCT.
In MF, hematopoietic engraftment after HCT may be delayed as a result of underlying BM fibrosis and hepatosplenomegaly. Moreover, because cytopenia is an AE of ruxolitinib, delayed engraftment or engraftment failure is a concern when ruxolitinib is administered during HCT. Our data show that addition of ruxolitinib is not associated with engraftment delay compared with our historical data previously reported by our group in this patient population.21 With the use of semiablative Flu/Mel conditioning, all patients achieved full/near-full donor chimerism early posttransplantation. In our study, mixed lymphoid and myeloid lineage was extremely rare, and with the small sample size, it was not possible to assess the impact of mixed chimerism on transplantation outcomes.
We observed promising rates of acute GVHD, with low incidence of grade 3 to 4 acute GVHD in our cohort of older adult patients (median age, 65 years), with a majority of HCTs (72%) from unrelated donors. Ruxolitinib has been shown to be effective as a therapeutic agent for established acute or chronic GVHD refractory to systemic corticosteroids.29-31 However, to date, ruxolitinib has not been prospectively evaluated as GVHD prophylaxis with Sir/Tac in clinical trials, and our data represent the first of such data, demonstrating the safety profile, pharmacokinetics, and clinical outcomes with major relevance in the evolving field of GVHD prevention and treatment. We previously evaluated and reported outcomes of patients with myelofibrosis (n = 110) who underwent allogeneic HCT with a Flu/Mel conditioning regimen at COH from 2004 to 201721 and reported cumulative incidence of grade 2 to 4 and 3 to 4 acute GVHD by day 100 of 45% and 17%, respectively. Cumulative incidence of chronic extensive GVHD at 12 months was 45%. In the current study, peri-HCT administration of ruxolitinib resulted in lower rates of grade 2 to 4 and 3 to 4 acute GVHD of 17% and 11%, respectively. Ruxolitinib administration lowered the cumulative incidence of moderate/severe chronic GVHD; at 1 year, the rate was 24%.
Because of the small sample size, it was not possible to definitively demonstrate the dose-dependent differences in cellular immune reconstitution or serum cytokine levels between DL1 and DL2 of ruxolitinib. Interestingly, our exploratory analyses without adjustment for multiple testing showed potential dose relation in Th1 cytokines levels (ie, IL-2, IFN-γ, and TNF-α) consistent with the proposed mechanisms of action32 and supporting that ruxolitinib at 10 mg twice daily is potentially more biologically active and immunologically favorable than 5 mg twice daily. The difference in levels of some of these cytokines (IL-2, IFN-γ, and TNF-α) persisted at day 100, even though ruxolitinib was stopped at day 33. This may mean that a shorter course of ruxolitinib has lasting effects on the cytokine milieu even after it is stopped.
In summary, in this pilot phase 1 trial, we successfully established the safety, feasibility, and RP2D of ruxolitinib during the peri-HCT period in patients with MF undergoing HCT, which was also associated with preventing severe acute GVHD. Future studies to optimize the duration and tapering strategies of ruxolitinib are of importance, and our data support a larger phase 2 randomized trial to prospectively define the benefits of ruxolitinib in the peritransplantation setting.
Acknowledgments
The authors thank City of Hope staff and nurses, as well as the patients and their families, without whom this work would not be possible. The authors also thank Golnaz Namdar and Maridel Blandino for their hard work and Arnab Chowdhury for his help with creating cytokine analysis plots.
This pilot trial was funded by an intramural chairs discretionary grant and National Cancer Institute, National Institutes of Health grant P30 CA033572 (Biostatistics and Analytical Pharmacology Core).
Authorship
Contribution: H.A., D.S.S., and R.N. contributed to study concept and design and data interpretation; N.-C.T., T.S., and J.P. were the study biostatisticians and performed statistical analysis; T.S. designed and performed pharmacokinetic studies and analysis; W.T. performed flow cytometry and cytokine analysis experiments; S.M. drafted the report; the remaining authors contributed to critical revision of the manuscript for intellectual content; and all authors read and approved the final version.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Haris Ali, Department of Hematology/HCT, 1500 E. Duarte Rd, Duarte, CA 91010; e-mail: harisali@coh.org.