

Key Points
Mutations affecting NF-κB, epigenomic regulation, or DNA damage repair were identified in MYD88 wild-type WM.
NF-κB pathway mutations were downstream of BTK, and many overlapped with those found in aggressive B-cell lymphomas.

Abstract
Activating MYD88 mutations are present in 95% of Waldenström macroglobulinemia (WM) patients, and trigger NF-κB through BTK and IRAK. The BTK inhibitor ibrutinib is active in MYD88-mutated (MYD88MUT) WM patients, but shows lower activity in MYD88 wild-type (MYD88WT) disease. MYD88WT patients also show shorter overall survival, and increased risk of disease transformation in some series. The genomic basis for these findings remains to be clarified. We performed whole exome and transcriptome sequencing of sorted tumor samples from 18 MYD88WT patients and compared findings with WM patients with MYD88MUT disease. We identified somatic mutations predicted to activate NF-κB (TBL1XR1, PTPN13, MALT1, BCL10, NFKB2, NFKBIB, NFKBIZ, and UDRL1F), impart epigenomic dysregulation (KMT2D, KMT2C, and KDM6A), or impair DNA damage repair (TP53, ATM, and TRRAP). Predicted NF-κB activating mutations were downstream of BTK and IRAK, and many overlapped with somatic mutations found in diffuse large B-cell lymphoma. A distinctive transcriptional profile in MYD88WT WM was identified, although most differentially expressed genes overlapped with MYD88MUT WM consistent with the many clinical and morphological characteristics that are shared by these WM subgroups. Overall survival was adversely affected by mutations in DNA damage response in MYD88WT WM patients. The findings depict genomic and transcriptional events associated with MYD88WT WM and provide mechanistic insights for disease transformation, decreased ibrutinib activity, and novel drug approaches for this population.
Introduction
Activating MYD88 and CXCR4 activations are present in 95% to 97% and 35% to 40% of Waldenström macroglobulinemia (WM) patients, respectively.1 Among WM patients who harbor an MYD88 mutation (MYD88MUT), nearly all carry the amino acid substitution p.Leu265Pro., making the identification of this mutation an important part of the diagnostic workup of WM.2 At the protein level, MYD88MUT triggers NF-κB pro-survival signaling through BTK and IRAK4/IRAK1, and activates the SRC family member HCK that triggers BTK, AKT, and ERK1/2 signaling.3,4 Ibrutinib blocks BTK and HCK activity and is highly active in MYD88MUT, but less so in MYD88WT WM, suggesting important differences in the molecular pro-survival signaling for these 2 WM variants.5-7 In some series, those with MYD88WT disease also showed increased risk of transformation to diffuse large B-cell lymphoma (DLBCL) and/or decreased overall survival (OS).8-10 CXCR4 mutations that impact bone marrow (BM) disease burden, immunoglobulin M (IgM) secretion, symptomatic hyperviscosity, and drug resistance are nearly always found in those patients with MYD88MUT WM.1,8,11,12 Although these findings allude to important biological differences between MYD88MUT and MYD88WT disease, the underlying genomic and transcriptional landscape of MYD88WT WM remains to be clarified. We therefore performed whole exome sequencing (WES) and transcriptome sequencing of the MYD88WT WM and compared the findings with MYD88MUT WM.
Patients and methods
The study was approved by Dana Farber/Harvard Cancer Center institutional review board, and patients provided written consent. Lymphoplasmacytic cells were collected by CD19+ MACS micro-bead selection (Miltenyi-Biotech, Auburn, CA) from BM aspirates of 18 consecutive patients meeting clinicopathological criteria for WM and MYD88WT disease following allele-specific polymerase chain reaction for detection of MYD88 L265P mutations and Sanger sequencing to exclude non-L265P MYD88 mutations.6,13 Baseline clinical information is shown in Table 1. For 12 patients, CD19-depleted peripheral blood mononuclear cells were available and used to prepare germline DNA as before.13-15 Fifty base-pair paired-end RNA sequencing libraries were generated using NEBNext Ultra RNA library prep kit (New England BioLabs, Ipswich, MA). WES libraries were constructed using SureSelect (Agilent, Santa Clara, CA) for 150 base pair paired-end sequencing. For tumor/germline paired samples, small variants were analyzed using both Strelka (https://github.com/Illumina/strelka) and MuTect2 (https://software.broadinstitute.org/gatk/). Unpaired WM samples were analyzed by GATK HaplotypeCaller (https://software.broadinstitute.org/gatk/). Mutations in unpaired patients were assessed for genes known to be relevant to WM and/or related lymphomas. Mutations were filtered for those that affected amino acid coding and were present in a gene that had measurable gene expression in healthy donor and/or WM samples. Somatic structural variants were detected using Manta (https://github.com/Illumina/manta); copy number alterations were called using Control-FREEC (http://boevalab.com/FREEC/). Variants were annotated using the Variant Effect Predictor (https://github.com/Ensembl/ensembl-vep). RNA sequencing reads were aligned using STAR (https://github.com/alexdobin/STAR) and quantified using Salmon (https://combine-lab.github.io/salmon/). Statistical analysis was performed using R, and Bioconductor packages limma, edgeR, and tximport were used to calculated voom-based differential gene expression testing. The DESeq2 package was used for regularized log transformation for clustering analysis and camera was used to calculate gene set enrichment using the publicly available MSigDB data set (http://bioinf.wehi.edu.au/software/MSigDB/). Sequencing data have been applied for deposition in the National Center for Biotechnology Information’s Short Read Archive. Results were compared with our previous genome, transcriptome, and OS findings for MYD88MUT WM patients.9,14-16 The survival from WM diagnosis, defined as the time between WM diagnosis to last follow-up or death, was estimated using the Kaplan-Meier method. All reported P values have been adjusted using the false discovery rate correction when appropriate.
Patient clinical characteristics
| Median | Range or % | |
|---|---|---|
| Age, y | 59 | 42-81 |
| Sex | 10 males/8 females | NA |
| BM, % | 12.5 | 2.5-80 |
| sIgM, mg/dL | 2625 | 610-5620 |
| Hb, g/dL | 11.0 | 8.1-14.4 |
| Adenopathy | 9 (50%) | NA |
| Splenomegaly | 7 (38.8%) | NA |
| Prior therapies | 1 | 0-4 |
| Untreated, n | 8 | 44.4% |
| Previously treated, n | 10 | 55.5% |
| Rituximab monotherapy | 2 | 20.0% |
| Alkylators | 7 | 70.0% |
| Nucleoside analogs | 5 | 50.0% |
| Proteasome inhibitors | 5 | 50.0% |
| Median | Range or % | |
|---|---|---|
| Age, y | 59 | 42-81 |
| Sex | 10 males/8 females | NA |
| BM, % | 12.5 | 2.5-80 |
| sIgM, mg/dL | 2625 | 610-5620 |
| Hb, g/dL | 11.0 | 8.1-14.4 |
| Adenopathy | 9 (50%) | NA |
| Splenomegaly | 7 (38.8%) | NA |
| Prior therapies | 1 | 0-4 |
| Untreated, n | 8 | 44.4% |
| Previously treated, n | 10 | 55.5% |
| Rituximab monotherapy | 2 | 20.0% |
| Alkylators | 7 | 70.0% |
| Nucleoside analogs | 5 | 50.0% |
| Proteasome inhibitors | 5 | 50.0% |
Hb, hemoglobin; NA, not available; sIgM, serum IgM.
Coded deidentified samples were collected under an approved sample collection protocol, institutional review board number 07-150.
Results
A median of >90.9 (range, 62.6-137.4) million reads were successfully mapped and paired following WES. Removing multimapping and duplicate reads resulted a median coverage of 157 (range, 96-230) reads per base pair over the target regions. Aligned data files were further analyzed with ContEst (http://www.broadinstitute.org/cancer/cga/contest) revealing minimal sample cross contamination with median estimated contamination levels of 0.05% (range, 0.02%-0.11%). The median number of somatic mutations per patient was 33 (range, 8-294; Figure 1A). Somatic variants for MYD88WT patients fell into 3 broad categories and included those predicted to (1) trigger NF-κB; (2) impart epigenomic dysregulation; and (3) impair DNA damage repair (DDR). The key mutation findings and predicted protein changes are shown in Table 2. A complete list of variants is reported in supplemental Table 1. Mutations predicted to activate NF-κB were observed in 12/18 (66.7%) patients and included TBL1XR1, PTPN13, MALT1, BCL10, NFKB1, NKFB2, NFKBIB, NFKBIZ, and UDRL1F (Figure 1B). Although many of these variants were previously identified in patients with aggressive B-cell lymphomas, novel recurring mutations also emerged.17-19 TBL1XR1 mutations that are also found in DLBCL and primary central nervous system lymphoma were identified in 5 (28%) MYD88WT patients, and included missense, nonsense, and frameshift mutations. Two patients each harbored 2 different TBL1XR1 mutations. TBL1XR1 mutations occurred at sites within or proximal to WD40 domains (Figure 1C) that are known to trigger TBL1XR1/nuclear receptor corepressor binding and degradation of nuclear receptor corepressor leading to activation of NF-κB and JUN pro-survival signaling.20
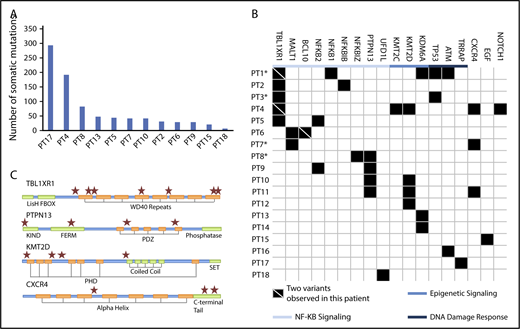
Mutations identified in MYD88WTWM by whole exome sequencing. (A) The median number of somatic mutations for patients with paired tumor/germline samples was 33 and the number of mutations per patient for these individuals are shown. (B) Somatic mutations were associated with NF-κB signaling, epigenetic regulation, and DNA damage response. Each row represents a unique patient. Patient identifiers in bold type indicate that the patient is deceased. *Patients with disease that later transformed. (C) Location of conserved motifs in the protein coding domains of top affected genes are shown. ★Location of a somatic mutation.
Mutations identified in MYD88WTWM by whole exome sequencing. (A) The median number of somatic mutations for patients with paired tumor/germline samples was 33 and the number of mutations per patient for these individuals are shown. (B) Somatic mutations were associated with NF-κB signaling, epigenetic regulation, and DNA damage response. Each row represents a unique patient. Patient identifiers in bold type indicate that the patient is deceased. *Patients with disease that later transformed. (C) Location of conserved motifs in the protein coding domains of top affected genes are shown. ★Location of a somatic mutation.
Observed somatic mutations in MYD88WT WM
| Gene | Consequence | Chr | Position | Variant | Protein | COSMIC | CADD |
|---|---|---|---|---|---|---|---|
| BCL10 | Nonsense | 1 | 85733609 | T/A | p.135R>* | COSM220638 | 38 |
| BCL10 | Frameshift | 1 | 85733357-8 | −/AGAGTTTGCACAAG | p.-/218-219LVQTX | NA | |
| CXCR4 | Deletion | 2 | 136872441-82 | TCTGTTTCCACTGAGTC TGAGTCTTCAAGTTTT CACTCCAGCTaa/taa | p.SVSTESESSSFH SS*339-353* | NA | |
| CXCR4 | Frameshift | 2 | 136872566-7 | −/T | p.T315NX | NA | |
| CXCR4 | Missense | 2 | 136873098 | G/T | p.R134S | 26.9 | |
| NFKBIZ | Missense | 3 | 101574709 | A/C | p.K45T | 26.3 | |
| TBL1XR1 | Missense | 3 | 176743302 | A/G | p.510L>S | 26.1 | |
| TBL1XR1 | Missense | 3 | 176744171 | G/A | p.S503L | COSM5000343 | 34 |
| TBL1XR1 | Splice acceptor | 3 | 176750925 | C/G | NA | 25.8 | |
| TBL1XR1 | Deletion | 3 | 176756175-7 | AAG/− | p.SC324-325C | COSM3205534 | NA |
| TBL1XR1 | Nonsense | 3 | 176767829 | G/A | p.Q220* | 39 | |
| TBL1XR1 | Missense | 3 | 176768267 | C/G | p.G187GR | 33 | |
| TBL1XR1 | Frameshift | 3 | 176769342 | T/− | p.N126NX | COSM1420706 | 34 |
| PTPN13 | Missense | 4 | 87556423 | T/A | p.L5Q | 33 | |
| PTPN13 | Missense | 4 | 87656789 | G/T | p.A732S | COSM5019859 | 27.6 |
| PTPN13 | Missense | 4 | 87683919 | A/C | p.N1198T | 3.649 | |
| PTPN13 | Missense | 4 | 87696460 | C/A | p.P1882Q | COSM481650 | 25.4 |
| NFKB1 | Missense | 4 | 103459060 | G/A | p.G69R | 31 | |
| KMT2C | Nonsense | 7 | 151891205 | C/A | p.G1517* | COSM3304224 | 41 |
| NOTCH1 | Nonsense | 9 | 139390945 | G/A | p.Q2416* | COSM4775108 | 41 |
| NFKB2 | Deletion | 10 | 104160996-1855 | NA | NA | NA | |
| NFKB2 | Deletion | 10 | 104160849-1688 | NA | NA | NA | |
| ATM | Nonsense | 11 | 108175504 | C/T | p.Q1867* | 37 | |
| ATM | Missense | 11 | 108204685 | T/C | p.M2667T | 26.3 | |
| KMT2D | Frameshift | 12 | 49433373-4 | −/G CCG CCCCCCT | p.-2691- | NA | |
| 2692AAPX | |||||||
| KMT2D | Missense | 12 | 49445543 | T/G | p.E641D | 5.499 | |
| KMT2D | Missense | 12 | 49446710 | G/T | p.P367Q | 11.92 | |
| KMT2D | Frameshift | 12 | 49448408 | G/− | p.G101X' | NA | |
| TP53 | Missense | 17 | 7577108 | C/A | p.C277F | COSM562338 | 34 |
| TP53 | Missense | 17 | 7577114 | C/A | p.C275F | COSM99932 | 34 |
| MALT1 | Nonsense | 18 | 56414859 | C/T | p.Q743* | 36 | |
| MALT1 | Nonsense | 18 | 56414882 | C/A | p.Y750* | 36 | |
| NFKBIB | Frameshift | 19 | 39398226-7 | CT/− | p.P299X | COSM5081722 | NA |
| UFD1L | Missense | 22 | 19443248 | C/A | p.G145V | 23.6 | |
| KDM6A | Missense | X | 44911044 | T/A | p.L249I | 25.4 | |
| KDM6A | Frameshift | X | 44942757 | G/− | p.V1113X | COSM5031082 | NA |
| KDM6A | Frameshift | X | 44922936-7 | −/GGAAGTGGAAGT | p-/.599- | NA | |
| AAT GGAAAC GTGCC | 600GSGSNGNVX |
| Gene | Consequence | Chr | Position | Variant | Protein | COSMIC | CADD |
|---|---|---|---|---|---|---|---|
| BCL10 | Nonsense | 1 | 85733609 | T/A | p.135R>* | COSM220638 | 38 |
| BCL10 | Frameshift | 1 | 85733357-8 | −/AGAGTTTGCACAAG | p.-/218-219LVQTX | NA | |
| CXCR4 | Deletion | 2 | 136872441-82 | TCTGTTTCCACTGAGTC TGAGTCTTCAAGTTTT CACTCCAGCTaa/taa | p.SVSTESESSSFH SS*339-353* | NA | |
| CXCR4 | Frameshift | 2 | 136872566-7 | −/T | p.T315NX | NA | |
| CXCR4 | Missense | 2 | 136873098 | G/T | p.R134S | 26.9 | |
| NFKBIZ | Missense | 3 | 101574709 | A/C | p.K45T | 26.3 | |
| TBL1XR1 | Missense | 3 | 176743302 | A/G | p.510L>S | 26.1 | |
| TBL1XR1 | Missense | 3 | 176744171 | G/A | p.S503L | COSM5000343 | 34 |
| TBL1XR1 | Splice acceptor | 3 | 176750925 | C/G | NA | 25.8 | |
| TBL1XR1 | Deletion | 3 | 176756175-7 | AAG/− | p.SC324-325C | COSM3205534 | NA |
| TBL1XR1 | Nonsense | 3 | 176767829 | G/A | p.Q220* | 39 | |
| TBL1XR1 | Missense | 3 | 176768267 | C/G | p.G187GR | 33 | |
| TBL1XR1 | Frameshift | 3 | 176769342 | T/− | p.N126NX | COSM1420706 | 34 |
| PTPN13 | Missense | 4 | 87556423 | T/A | p.L5Q | 33 | |
| PTPN13 | Missense | 4 | 87656789 | G/T | p.A732S | COSM5019859 | 27.6 |
| PTPN13 | Missense | 4 | 87683919 | A/C | p.N1198T | 3.649 | |
| PTPN13 | Missense | 4 | 87696460 | C/A | p.P1882Q | COSM481650 | 25.4 |
| NFKB1 | Missense | 4 | 103459060 | G/A | p.G69R | 31 | |
| KMT2C | Nonsense | 7 | 151891205 | C/A | p.G1517* | COSM3304224 | 41 |
| NOTCH1 | Nonsense | 9 | 139390945 | G/A | p.Q2416* | COSM4775108 | 41 |
| NFKB2 | Deletion | 10 | 104160996-1855 | NA | NA | NA | |
| NFKB2 | Deletion | 10 | 104160849-1688 | NA | NA | NA | |
| ATM | Nonsense | 11 | 108175504 | C/T | p.Q1867* | 37 | |
| ATM | Missense | 11 | 108204685 | T/C | p.M2667T | 26.3 | |
| KMT2D | Frameshift | 12 | 49433373-4 | −/G CCG CCCCCCT | p.-2691- | NA | |
| 2692AAPX | |||||||
| KMT2D | Missense | 12 | 49445543 | T/G | p.E641D | 5.499 | |
| KMT2D | Missense | 12 | 49446710 | G/T | p.P367Q | 11.92 | |
| KMT2D | Frameshift | 12 | 49448408 | G/− | p.G101X' | NA | |
| TP53 | Missense | 17 | 7577108 | C/A | p.C277F | COSM562338 | 34 |
| TP53 | Missense | 17 | 7577114 | C/A | p.C275F | COSM99932 | 34 |
| MALT1 | Nonsense | 18 | 56414859 | C/T | p.Q743* | 36 | |
| MALT1 | Nonsense | 18 | 56414882 | C/A | p.Y750* | 36 | |
| NFKBIB | Frameshift | 19 | 39398226-7 | CT/− | p.P299X | COSM5081722 | NA |
| UFD1L | Missense | 22 | 19443248 | C/A | p.G145V | 23.6 | |
| KDM6A | Missense | X | 44911044 | T/A | p.L249I | 25.4 | |
| KDM6A | Frameshift | X | 44942757 | G/− | p.V1113X | COSM5031082 | NA |
| KDM6A | Frameshift | X | 44922936-7 | −/GGAAGTGGAAGT | p-/.599- | NA | |
| AAT GGAAAC GTGCC | 600GSGSNGNVX |
CADD, combined annotation dependent depletion; chr, chromosome; COSMIC, Catalogue of Somatic Mutations in Cancer.
Somatic mutations in the phosphatase PTPN13 were observed in 4 (22%) patients, occurring within the PDZ, FERM, and KIND domains (Figure 1C). The PDZ domain binds to IKBA, an essential cytosolic gatekeeper of NF-κB.21 Loss of PTPN13 function leads to tyrosine phosphorylation of IKBA, resulting in nuclear translocation of NF-κB. Other mutations predicted to alter NF-κB signaling included those in the CBM complex (MALT1, BCL10) in 3 (17%), and NFKB2 in 2 (11%) patients, and NFKB1, NFKBIB, NFKBIZ, and UFD1L, which were observed once. The 2 MALT1 variants were nonsense mutations 23 base pairs apart and predicted for truncation of the C-terminal domain with loss of a TRAF6-binding site. Mutations at this site have not been previously reported, although functional studies suggest a critical role in preventing MALT1 degradation and stabilizing the CBM complex.22 One patient carried both nonsense and frameshift mutations in the C-terminal domain of BCL10; these are similar to those in MALT and follicular lymphomas that abrogate pro-apoptotic activity and promote NF-κB activation.23 Structural variant analysis also revealed deletions removing the DEATH/PEST domain of NFKB2 in 2 patients covering amino acids 691 through 822 and 711 through 839, respectively. Deletions in this region are associated with constitutive NF-κB activation in myeloma.24
Somatic mutations in the chromatin-modifying genes (CMG) KMT2D, KDM6A, and KMT2C were also observed in 4 (22%), 3 (17%), and 1 (6%) of the MYD88WT WM patients, respectively. Mutations in the H3 lysine 4 methyltransferases KMT2D and KMT2C are commonly found in DLBCL and follicular non-Hodgkin lymphoma patients.17,18,25 Knockout studies have suggested a partial functional redundancy for these CMG.25 In KMT2D murine knockout models, reduced class-switched B cells were observed following immunization, a finding consistent with defective B-cell maturation and/or class switching.25 Mutations in the DDR genes TP53 2/18 (11%), ATM 1/18 (6%), and TRRAP 1/18 (6%) were also observed, and the TRRAP-mutated patient (patient 17) exhibited the highest number of somatic variants in this series (Figure 1A). A role for these mutations in NF-κB–driven lymphomagenesis is supported by previous functional studies, along with high rates of somatic mutations in TRRAP-mutated patients.26-28
Other mutations included CXCR4 in 3/18 (17%) patients (Figure 1C), 2 of whom had frameshift mutations within the C-terminal domain as those found in MYD88MUT WM patients.11 A third mutation (R134S) was identified in the intracellular 2 domain; these mutations have not been previously reported in CXCR4-mutated WM patients. Substitutions at R134 have been demonstrated to affect inhibitory G protein alpha subunit (Gai) activation.29 In addition, 1 NOTCH1 and 1 EGF mutation were observed. Analysis of copy number alterations revealed no recurring events, but was remarkable for the absence of chromosome 6q deletions that are present in half of MYD88-mutated WM patients, and target genes regulating BTK, BCL2, NF-κB, and apoptosis signaling.30 To better understand the relevance of these mutations in relationship to MYD88 mutation status, we compared the WES findings from this study with those from our previous whole genome sequencing of 53 MYD88MUT WM patients.14,15 Although many of the mutated genes in MYD88WT patients were also found in MYD88MUT patients, TBL1XR1 and MALT1 mutations were observed in MYD88WT patients only (P = .001 and 0.062, respectively), whereas those with KDM6A (P = .052) and KMT2D (P = .065) showed a trend toward enrichment in MYD88WT patients (Figure 2A).
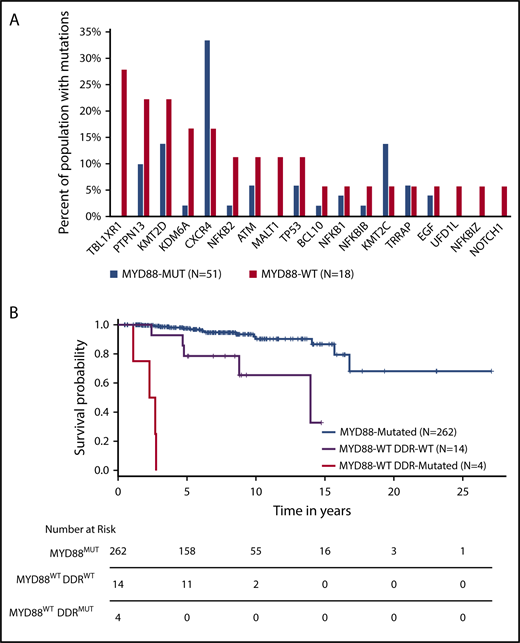
Comparison of findings for MYD88WTand MYD88MUTWM. Comparison of somatic mutation frequencies between MYD88WT and MYD88MUT WM patients. (A) Data for mutation frequencies for 53 MYD88MUT WM patients were acquired from our previous whole genome sequencing results, using high-quality somatic variants supported by at least 3 reads.10,11 (B) Kaplan-Meier curves for overall survival from time of diagnosis for WM patients with MYD88MUT, and MYD88WT with and without DDR mutations (log-rank P < .0001).
Comparison of findings for MYD88WTand MYD88MUTWM. Comparison of somatic mutation frequencies between MYD88WT and MYD88MUT WM patients. (A) Data for mutation frequencies for 53 MYD88MUT WM patients were acquired from our previous whole genome sequencing results, using high-quality somatic variants supported by at least 3 reads.10,11 (B) Kaplan-Meier curves for overall survival from time of diagnosis for WM patients with MYD88MUT, and MYD88WT with and without DDR mutations (log-rank P < .0001).
With a median follow-up of 72.1 months (range, 13.2-176.9) from diagnosis, 4 (22.2%) patients transformed to DLBCL. Nine (50%) died, including 3 from disease transformation. The genomic mutations found in transformed patients included TBL1XR1, TP53, NFKB1, NFKB2, and MALT1 somatic mutations, all of which have been identified in DLBCL patients.17,18 MYD88WT patients had a significantly lower median OS relative to patients with MYD88MUT disease. The estimated median OS for the 18 MYD88WT patients was 167 months; a median follow-up of 73.8 months was insufficient to calculate the predicted median for the cohort of 262 MYD88MUT patients diagnosed over the same period (log-rank P < .0001). Genomic findings were aggregated into NF-κB signaling, epigenetic signaling, and DDR categories and evaluated for their effect on OS. Particularly striking was the exceedingly poor survival in patients with DDR mutations, in whom the median OS was 29.9 months (range, 13.2-33.1), as shown in Figure 2B. No significant differences in OS were observed when stratifying the MYD88WT population by the other 2 categories. Constructing a Cox proportional hazard model accounting for sex, age at diagnosis, MYD88 mutation status, and the presence of DDR mutations revealed hazard ratios of 8.5 and 77.9 for MYD88WTDDRWT and MYD88WTDDRMUT, respectively, relative to MYD88MUT WM patients (P < .001 for both comparisons).
Analysis of the MYD88WT WM transcriptome revealed a distinct transcriptional profile (Figure 3A). However, principal component analysis of the top 500 high-variance genes revealed a clustering of MYD88WT and MYD88MUT WM samples, regardless of CXCR4 mutation status that was distinct from healthy donor peripheral blood B, memory B, and plasma cells (Figure 3B). These findings were recapitulated in the supervised clustering of the top 100 most statistically significant differentially expressed genes between healthy donor memory B cells and MYD88WT WM samples, in which gene expression levels were very similar between all WM samples regardless of MYD88 and CXCR4 mutation status (Figure 3C). Likewise, the contrast between healthy donor memory B cells and MYD88WT samples found significant log2 fold change (LFC) overexpression of genes we had previously associated with WM, including DNTT (LFC, 12.4; P = .005), RAG1 (LFC, 8.1; P = .008), RAG2 (LFC, 10.0; P < .001), CXCL12 (LFC, 11.8; P = .002), VCAM1 (LFC, 10.6; P = .001), IGF1 (LFC, 7.0; P < .001), BMP3 (LFC, 7.0; P = .005), CD5L (LFC, 10.0; P = .002), and B2M (LFC, 1.1; P = .022).15 These findings are likely to explain many of the shared clinical and morphological characteristics among WM patients, regardless of their underlying MYD88 mutation status. The exceptions were CXCR4, BCL2, and BAX, which were not significantly different from healthy donor controls in MYD88WT samples.
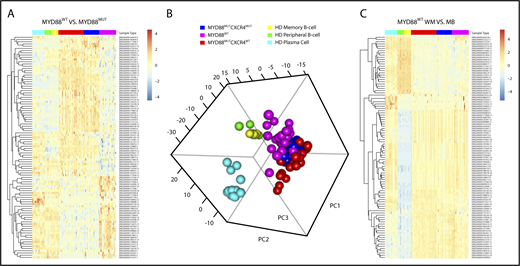
Findings from next-generation gene expression studies in MYD88WTWM. (A) The top 100 most statistically significant genes between samples from 18 MYD88WT and 75 MYD88MUT patients are shown, demonstrating a uniform gene signature associated with the MYD88WT population. (B) Principal component analysis of the top 500 high variance genes revealed a clustering of MYD88WT and MYD88MUT WM samples, regardless of CXCR4 mutation status that was distinct from healthy donor peripheral blood B, memory B, and plasma cells. (C) These findings were also recapitulated in the supervised clustering of the top 100 most statistically significant differentially expressed genes between healthy donor memory B cells and MYD88WT WM samples, in which gene expression levels were very similar between all WM samples regardless of MYD88 and CXCR4 mutation status.
Findings from next-generation gene expression studies in MYD88WTWM. (A) The top 100 most statistically significant genes between samples from 18 MYD88WT and 75 MYD88MUT patients are shown, demonstrating a uniform gene signature associated with the MYD88WT population. (B) Principal component analysis of the top 500 high variance genes revealed a clustering of MYD88WT and MYD88MUT WM samples, regardless of CXCR4 mutation status that was distinct from healthy donor peripheral blood B, memory B, and plasma cells. (C) These findings were also recapitulated in the supervised clustering of the top 100 most statistically significant differentially expressed genes between healthy donor memory B cells and MYD88WT WM samples, in which gene expression levels were very similar between all WM samples regardless of MYD88 and CXCR4 mutation status.
Comparisons of gene expression based on MYD88 mutation status revealed 291 significantly dysregulated genes that can be seen in supplemental Table 2. Many of the genes we previously associated with MYD88WT WM were validated in this larger cohort including IL6 (LFC, −3.7; P = .022), TNFAIP3 (LFC, −1.5; P = .04), NFKBIZ (LFC, −1.8; P = .034), PIM1 (LFC, −2.1; P < .001), PIM2 (LFC, −1.4; P = .038), CD40 (LFC, −1.4; P = .037), and CD86 (LFC, 2.7; P = .024). A significant dysregulation in a number of highly relevant novel genes including RASSF6 (LFC, −6.1; P = .02), EIF5A2 (LFC, 2.4; P = .008), CCL22 (LFC, −4.5; P = .034), CCR7 (LFC, −2.7; P = .006), LTK (LFC, 2.0; P = .028), VEGFA (LFC, −2.7; P = .027), PRDM8 (LFC, −2.6; P < .001), PRDM1 (LFC, −3.0; P = .001), and XBP1 (LFC, −1.9; P = .028) was also found in this expanded cohort. Gene set enrichment analysis identified significant enrichment for the upregulation of E2F, MYC, PIK3-AKT-MTOR, and G2M checkpoint signaling targets (P ≤ .009 for all) as well as the downregulation of inflammatory response genes (P = .023) and TNFA signaling through NF-κB (P < .001).
Discussion
This is the first study to focus on the genome and transcriptome of MYD88WT WM, an infrequent subtype of WM that is remarkable in certain studies for an increased risk of disease transformation, lower response to ibrutinib, and shortened OS.5-10,31 Distinct patterns of mutations were identified among MYD88WT patients, including those affecting NF-κB signaling, epigenomic regulators, and those in DDR genes, and were independent of prior treatment status. The most common mutations involved those affecting genes in NF-κB signaling that were identified in 12/18 (66.7%) MYD88WT patients, and included TBL1XR1, NFKBIB, NFKBIZ, NFKB2, MALT1, BCL10, and UDRLIF. Although mutations in these genes are rare or absent in MYD88MUT WM disease, they are found in aggressive lymphomas.17-19 TBL1XR1 mutations that were identified in 5 patients, including 2 patients who each had 2 mutations that are of particular interest given their frequent presence in activated B cell–like DLBCL and primary central nervous system lymphoma.13-15 These diseases are also recognized for their high frequency of recurring MYD88 mutations that are exclusive of TBL1XR1 mutations, suggesting that the actions of the latter may mimic at least in part those of activating MYD88 mutations.13-15 In addition to mutations in TBL1XR1, many of the other NF-κB pathway mutations identified in this study are found in aggressive B-cell lymphomas. Taken together, these findings may provide a genomic explanation for the increased risk of disease transformation9,10 and accompanying shorter survival observed in our previous study for MYD88WT WM patients.9 Somatic mutations in CMG were also observed in 8 (44.4%) MYD88WT patients. KMT2D mutations were the most common CMG mutations observed in MYD88WT WM patients, and are present in 30% of DLBCL patients. Varettoni et al32 recently reported KMT2D mutations in 24% of MYD88MUT WM patients, although these were primarily subclonal and their clinical course relative to patients without KMT2D mutations was not clarified. The mechanistic pathways by which CMG mutations promote WM pro-survival signaling deserves further study given their frequent occurrence in WM.
Particularly concerning were MYD88WT patients who presented with DDR mutations. Compared with patients with MYD88MUT and MYD88WT disease lacking DDR mutations, those with MYD88WT disease with DDR mutations represented a subset with ultra-high-risk disease. A similar observation has also been made in myeloma patients.33 Although TP53 mutations are uncommon in WM, they are present in MYD88-mutated patients.32,34,35 Their association with poor outcome in MYD88-mutated patients has previously been reported.34,35 Last, CXCR4 activating mutations found in 30% to 40% of MYD88MUT patients were identified in MYD88WT patients, although the frequency of these mutations was lower. Only 2 (9%) of the MYD88WT patients had C-terminal variants that promote WHIM-like signaling, as found in MYD88MUT WM patients. The significance of a third CXCR4 variant (R134S) identified in 1 patient remains unclear. All 3 of these CXCR4-mutated patients also had mutations affecting NF-κB signaling, akin to MYD88-mutated WM patients, and may therefore be amenable to therapeutics targeting CXCR4, such as ulocuplumab, which is being investigated in WM patients harboring both MYD88 and CXCR4 mutations in combination with ibrutinib (NCT03225716).
The findings of this study may also provide important insights into why WM patients with MYD88WT disease are less responsive to ibrutinib monotherapy.5-7 The NF-κB pathway mutations observed in two-thirds of MYD88WT patients were all downstream of BTK (Figure 4). NF-κB pathway inhibitors that are downstream of BTK, including proteasome inhibitors that target IKBA, and novel agents that target IKK and MALT1 may be more appropriate for these individuals.36,37 A mechanistic rationale for how ibrutinib fits into the treatment of CMG and DDR-mutated MYD88WT WM remains elusive, as does a targeted treatment approach for such patients.
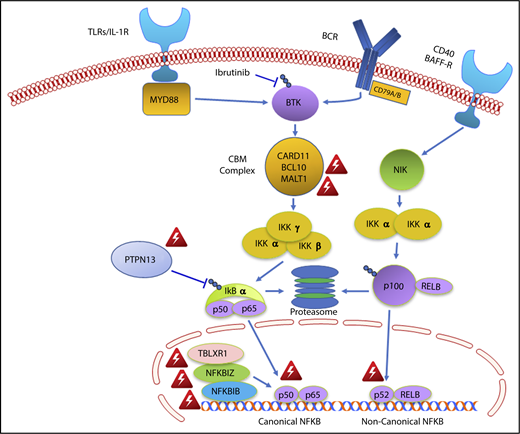
Genomic variants identified in MYD88 wild-type WM that affect NF-κB signaling. Red triangle denotes variants identified by whole exome sequencing in MYD88 wild-type WM patients.
Genomic variants identified in MYD88 wild-type WM that affect NF-κB signaling. Red triangle denotes variants identified by whole exome sequencing in MYD88 wild-type WM patients.
An unexpected finding was the transcriptional similarity for MYD88WT and MYD88MUT disease relative to healthy donor B cells. This finding may well account for the many overlapping disease characteristics observed between MYD88WT and MYD88MUT patients.8,9 The transcriptional similarity between these subsets of WM may reflect the common activation of NF-κB triggered by activating mutations such as TBL1XR1 in MYD88WT patients and mutated MYD88. However, the extent of NF-κB activation may differ, because some NF-κB–regulated genes such as IL6, IRAK2, TNFAIP3, NFKBIZ, NFKB2, TIRAP, PIM1, and PIM2 show lower expression in MYD88WT vs MYD88MUT patients. Because MYD88 is a key mediator of innate immune signaling, additional branch points for downstream signaling exist, even in the context of NF-κB that includes AKT and ERK (via cytokines) pathways triggered by MYD88 activation of HCK and/or BCR/SYK in WM cells.4,38 The existence of a “My-T-BCR supercomplex” that encompasses mutated MYD88 and BCR components that contribute to broader signaling that includes mTOR is also supported by recent studies in activated B cell–like DLBCL.39 Consistent with this notion, we observed a gene set enrichment for PI3K-AKT-MTOR signaling was observed in MYD88WT patients; therefore, a targeted approach for treating MYD88WT patients may entail the use of PI3K or MTOR inhibitors. In contrast to MYD88MUT patients, those with MYD88WT had lower levels of BCL2 expression that were on par with the expression found in healthy donor B cells. The BCL2 antagonist venetoclax has shown remarkable activity in WM, although MYD88 mutation status and relative dependence on BCL2 expression remain to be clarified.40
In summary, the findings depict genomic and transcriptional events associated with MYD88WT WM and provide mechanistic insights for disease transformation, decreased ibrutinib activity, and novel drug approaches for this population.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors acknowledge the contributions of Yaoyu Wang and John Quackenbush at the Center for Cancer Computational Biology at the Dana-Farber Cancer Institute for generating the next-generation sequencing data, as well as Mathew Temple and Leutz Buon of the Dana-Farber Research Computing Center for their assistance.
The work was supported by the Orszag Family Fund for WM Research, Peter S. Bing, International Waldenstrom’s Macroglobulinemia Foundation, Kerry Robertson Fund for WM, Leukemia and Lymphoma Society, National Institutes of Health, National Cancer Institute Development Award (grant Spore 5P50CA100707-12) (Z.R.H.), and an American Society of Hematology Scholar Award (Z.R.H.).
Authorship
Contribution: Z.R.H. and S.P.T. designed the study and wrote the manuscript; Z.R.H., M.K.S., and G.G.C. conducted the bioinformatic analysis; L.X., N.T., M.G.D., A.K., J.C., X.L., M.M., and G.Y., performed tumor cell isolation, and/or allele-specific polymerase chain reaction genotyping assays and Sanger sequencing; and S.P.T., J.J.C., C.J., K.C.A., N.C.M., C.J.P., K.M., J.G., and T.D. provided patient care, obtained samples, clinical data and/or analyzed clinical data.
Conflict-of-interest disclosure: Z.R.H., S.P.T., N.C.M., K.C.A., and J.J.C. have received consulting fees, and/or research funding from Pharmacyclics Inc., Janssen Inc., AbbVie Inc., and/or Bristol Myers Squibb. The remaining authors declare no competing financial interests.
Correspondence: Steven P. Treon, Bing Center for Waldenström’s Macroglobulinemia, Dana-Farber Cancer Institute, M547, 450 Brookline Ave, Boston MA 02215; e-mail: steven_treon@dfci.harvard.edu.

