


Abstract
Endothelial cell (EC) activation has been suspected of triggering a group of rare and dismal complications that can occur after allogeneic hematopoietic stem cell transplantation (HSCT). Capillary leak syndrome, engraftment syndrome, transplant-associated microangiopathy, diffuse alveolar hemorrhage, and idiopathic pneumonia syndrome are the main nosological entities. Post-HSCT endotheliitis can be triggered by chemotherapy, infections, and calcineurin inhibitors, but allogeneic reactivity is claimed to be the common denominator. Endothelial damages are thought to activate several deleterious pathways (proapoptotic, procoagulant, proinflammatory) and can lead to multiorgan failure; however, clinical manifestations of each syndrome overlap, and their relationship with graft-versus-host disease could be minimal. The lack of well-defined diagnostic criteria does not allow for a clear-cut comparison in the current literature. Therapeutic efforts have been made to intercept the pathogenic mechanisms leading to EC dysfunction, but remission rates and survival remain mostly unsatisfactory. In this article, we have reviewed the incidence, clinical features, and treatment approaches of EC activation syndromes, and we plead for the development of internationally accepted standard definitions.
Introduction
Hematopoietic stem cell transplantation (HSCT) is associated with early and late severe complications.1,2 Vascular endothelial syndromes are a range of life-threatening complications that often result in a sudden decline of a patient’s clinical conditions. The most recognized systemic entities include capillary leak syndrome (CLS), engraftment syndrome (ES), transplant-associated thrombotic microangiopathy (TA-TMA), and, in the lungs, idiopathic pneumonia syndrome (IPS).3-6
However, because the clinical manifestations may overlap between different clinical entities, the actual incidence of each form is mostly unknown. A variety of trigger factors may lead to these complications, including the toxicity of the conditioning regimen, various drugs, infections, and inflammation (including the allogeneic reaction).7-9 Their pathogenic correlation and their clinical overlap with graft-versus-host disease (GVHD) are often challenging. Indeed, the absence of well-defined diagnostic criteria and well-established treatments make the management of these syndromes a major practical issue for transplant physicians.
Veno-occlusive disease/sinusoidal obstruction syndrome (VOD/SOS) has long been considered an entity belonging to this spectrum.10 However, because its pathogenesis is not entirely endothelial, we believe that this syndrome should be considered a separate entity (see “Controversial issues and management strategies”).
In this review, we will focus on pathophysiological evidence, clinical characteristics, and the main issues concerning post-HSCT endothelial cell (EC) activation syndromes.
Pathophysiology
The term “EC activation” includes a broad spectrum of phenotypic changes in the endothelium. Capillary permeability is a tightly controlled feature of the microcirculation in all organ beds that becomes increased in inflammatory conditions, resulting in net extravasation of fluid out of the vascular space and into the tissues.11 The underlying pathogenic events are often shared, accounting for the close clinical association observed among endothelial syndromes, infections, and GVHD.12 When the activating stimulus is too intense or persistent, it can produce a localized or systemic dysfunction of ECs. The different denomination of these syndromes is determined by the predominant phenotypic change (proinflammatory, prothrombotic, proapoptotic) and its localization (systemic or organ related).11,13
The pathophysiological mechanism of EC activation is multifactorial and may involve prominent cellular interactions among T cells, monocytes, and other effector cells, together with complement activation and proinflammatory cytokine production and release.14
Experimental evidence suggests that alloreactivity per se plays a role in the pathogenesis of these endothelial complications.15-17 Several models showed that ECs are targets for alloreactive T lymphocytes in acute and chronic GVHD.15,18 This immune-mediated endothelial damage, together with the unspecific chronic vascular inflammation observed during GVHD, can have a role in the development of the endothelial complications observed after allogeneic HSCT, especially in the context of a concurrent clinical GVHD.19 Moreover, ECs are supposed to play a key role in immunoregulation during acute GVHD through the modulation of expression of several T helper 1 cell regulators (eg, T-cell immunoglobulin and mucin domain-3 ligand or galectin-9), which may be responsible for target organ damage in GVHD.20 The antigen-presenting capacity of ECs has been assessed in various in vivo and in vitro studies, and it seems to contribute to the initial stimulation of alloreactive T lymphocytes.21,22
The pathogenesis of endothelial syndromes may be dependent upon the intensity of the preparative regimen and the stem cell source.13 Endothelial injury from highly cytotoxic conditioning regimens induces proinflammatory cytokines (eg, interleukin-1 [IL-1], IL-8, IL-2, tumor necrosis factor-α [TNF-α], and interferon-γ [IFN-γ]), increased release of procoagulant factors (eg, von Willebrand factor, thrombomodulin, plasminogen activator inhibitor-1), and overexpression of soluble adhesion molecules (eg, soluble E-selectin [sE-selectin], sICAM-1, sVCAM-1). The effects of these soluble markers on subsequent cellular and cytokine interactions have been well established since the late 1990s, but results are not always consistent.13,23,24 In a more recent analysis, an association was found between acute GVHD–related biomarkers (eg, suppression of tumorigenicity 2, elafin, and regenerating islet-derived 3α) and EC activation markers (sVCAM-1 and plasminogen activator inhibitor-1), suggesting the link between these entities.16
A relevant role is also attributed to soluble adhesion molecules: serum levels of sVCAM-1, sE-selectin, and sICAM-1 are increased after HSCT.25,26
Two other markers have been described: microparticles (detected as particles shed from activated cells, such as platelets, monocytes, polymorphonuclear cells, and ECs) and circulating ECs, which seem to reflect the extent of endothelial damage in many disorders.27,28 Those markers have been found to be increased in patients with acute GVHD and thrombotic microangiopathy (TMA) but not during conditioning or sepsis.29
Thus, several soluble factors associated with endothelial function have been investigated in the context of HSCT, aiming to identify reliable biomarkers of endothelial complications after HSCT. However, even if some of them may have a potential role as indicators of endothelium activation/damage, they are not specific and are largely influenced by several host factors. Indeed, no consensual biomarker can be used for the diagnosis or monitoring of post-HSCT endothelial complications.
Clinical entities
Figure 1 summarizes some critical pathophysiological features of EC activation syndromes.
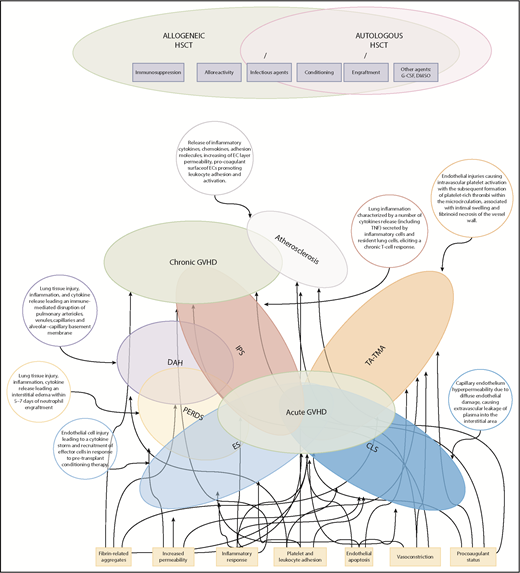
Spectrum and pathophysiology of post-HSCT endothelial complications. The Venn diagram highlights the overlap of endothelial syndromes, both diagnostically and pathophysiologically, with GVHD. Many clinical and pathophysiological features of GVHD parallel those of other EC activation syndromes in which a single organ is targeted. Here, atherosclerosis and several delayed forms of IPS have been proposed as late endothelial dysfunction syndromes overlapping with chronic GVHD. DAH, diffuse alveolar hemorrhage; PERDS, peri-engraftment respiratory distress syndrome.
Spectrum and pathophysiology of post-HSCT endothelial complications. The Venn diagram highlights the overlap of endothelial syndromes, both diagnostically and pathophysiologically, with GVHD. Many clinical and pathophysiological features of GVHD parallel those of other EC activation syndromes in which a single organ is targeted. Here, atherosclerosis and several delayed forms of IPS have been proposed as late endothelial dysfunction syndromes overlapping with chronic GVHD. DAH, diffuse alveolar hemorrhage; PERDS, peri-engraftment respiratory distress syndrome.
TA-TMA
TA-TMA is characterized by EC activation and thrombus formation. TA-TMA manifests itself with the triad of hemolytic anemia, thrombocytopenia, and organ failure and is associated with a high mortality.30 The reported incidence of TMA after HSCT varies significantly (from 0% to 74%), owing to the lack of an established definition.31 TA-TMA has been related to the conditioning regimen, total body irradiation, infections, and immunosuppressive therapy. The diagnosis is based on laboratory criteria associated with these conditions (ie, red cell fragmentation, anemia, increased lactose dehydrogenase (LDH), low haptoglobin, and thrombocytopenia).5,32
There are 4 proposed consensus criteria developed for TA-TMA5,32-34 (Table 1). In contrast to other TMAs, the pathophysiology of TA-TMA has not been fully elucidated, even if endothelium damage seems to play a major role, and the association with drugs (eg, calcineurin inhibitors [CNIs]), conditioning regimen, allogeneic immune reactions (eg, clinical GVHD or subtle allogeneic immune response), or infectious complications is well established.35-37
Diagnostic criteria for TA-TMA
| Clinical/laboratory finding | BMT-CTN criteria32 | IWG-EBMT criteria5 | Overall TMA criteria33 | Refined TMA criteria34 |
|---|---|---|---|---|
| Normal coagulation assays* | Yes | Yes | Yes | NR |
| Schistocytosis | >2 per HPF on PS | >8 per HPF on PS | >2 per HPF on PS | Yes |
| Increase in serum LDH | Yes | Yes | Yes | Yes |
| Thrombocytopenia† | NR | Yes | Yes | Yes |
| Anemia | NR | Yes | Yes | Yes |
| Decrease in serum haptoglobin | NR | Yes | Yes | NR |
| Negative Coombs test | Yes | NR | Yes | NR |
| Renal and/or CNS involvement | Yes | NR | NR | NR |
| Proteinuria | NR | NR | NR | Random urinalysis proteinuria concentration ≥30 mg/dL‡ |
| Hypertension | NR | NR | NR | 3-18 y: BP at 95th percentile value for age, sex, and height; >18 y: BP ≥140/90 mm Hg |
| Terminal complement assay | NR | NR | NR | Elevated plasma concentration of sC5b-9‡ |
| Clinical/laboratory finding | BMT-CTN criteria32 | IWG-EBMT criteria5 | Overall TMA criteria33 | Refined TMA criteria34 |
|---|---|---|---|---|
| Normal coagulation assays* | Yes | Yes | Yes | NR |
| Schistocytosis | >2 per HPF on PS | >8 per HPF on PS | >2 per HPF on PS | Yes |
| Increase in serum LDH | Yes | Yes | Yes | Yes |
| Thrombocytopenia† | NR | Yes | Yes | Yes |
| Anemia | NR | Yes | Yes | Yes |
| Decrease in serum haptoglobin | NR | Yes | Yes | NR |
| Negative Coombs test | Yes | NR | Yes | NR |
| Renal and/or CNS involvement | Yes | NR | NR | NR |
| Proteinuria | NR | NR | NR | Random urinalysis proteinuria concentration ≥30 mg/dL‡ |
| Hypertension | NR | NR | NR | 3-18 y: BP at 95th percentile value for age, sex, and height; >18 y: BP ≥140/90 mm Hg |
| Terminal complement assay | NR | NR | NR | Elevated plasma concentration of sC5b-9‡ |
Doubling of serum creatinine from baseline (baseline creatinine before hydration and conditioning) or 50% decrease in creatinine clearance from baseline.
BP, blood pressure; BMT-CTN, Blood and Marrow Transplant Clinical Trials Network; CNS, central nervous system; HPF, high-power field; IWG, International Working Group; LDH, lactose dehydrogenase; PS, peripheral smear.
Coagulation assays included prothrombin time and activated partial thromboplastin time.
De novo, prolonged, or progressive thrombocytopenia (platelet count 50 × 109/L or 50% reduction from previous counts).
Parameters of severity of TMA, according to Jodele et al.34
Clinical phenotype may overlap with that of other thrombotic microangiopathies, such as thrombotic thrombocytopenic purpura (TTP) or hemolytic uremic syndrome (HUS). In TTP and HUS, because of concurrent consumption in coagulation factors, patients tend to have more bleeding and macrovascular complications, including deep venous thrombosis and cerebral artery thrombosis,38 and the clinical presentation regularly involves central nervous system (CNS) and renal dysfunction, sparing organs such as the liver or the lungs. In TA-TMA, even if the kidneys and CNS can be typically affected, the endothelial damage is systemic and can affect other organs, including the lungs, gastrointestinal tract, and heart.39,40 Idiopathic TTP has been attributed to deficient activity of ADAMTS-13, a metalloproteinase responsible for cleaving ultralarge von Willebrand factor multimers.41 Severely deficient ADAMTS-13 activity (<5% of normal) is associated with TTP in 33% to 100% of patients.42,43 On the contrary, TA-TMA is usually associated with normal ADAMTS-13 activity (>5%). Evidence of complement activation was found in TA-TMA patients, as well as in HUS patients, and elevated sC5b-C9 is associated with prognosis.44,45 The mechanism of complement involvement has been investigated in detail. Neutralizing anti–complement factor H antibodies have been detected in some patients.34 Furthermore, a recent study reported germline mutation/polymorphisms in complement-related genes in more than half of TA-TMA patients.46 These pathogenic insights are seminal, because therapeutic complement inhibitors are available and may be used for the etiologic treatment of TA-TMA.47-49
Lung endothelial injury
IPS is a post-HSCT lung disease that is characterized by signs and symptoms of pneumonia and evidence of widespread alveolar injury, in which infectious etiologies and cardiac dysfunction, acute renal failure, or iatrogenic fluid overload have been excluded, according to the American Thoracic Society definition.6 Its incidence in the first 120 days after HSCT has been estimated at ∼5% to 8% following myeloablative conditioning and at 2% after nonmyeloablative conditioning. Its mortality is very high, ranging from 60% to 80%, and it is >95% for patients requiring mechanical ventilation.50,51 Median time of onset is generally 19 days (range: 4-106) after HSCT.50 This lung injury may occur after autologous or allogeneic HSCT. After autologous HSCT, IPS usually has a favorable prognosis and responds to corticosteroid therapy. IPS after allogeneic HSCT is associated with a poor response to corticosteroids and progressive respiratory failure.
Different risk factors have been identified: the intensity of conditioning regimens, concomitant acute GVHD, older recipient age, malignancy other than leukemia, HLA mismatch, and use of methotrexate and busulfan.50 Diagnostic criteria for IPS include signs and symptoms of pneumonia, multilobar radiographic infiltrates, abnormal pulmonary function, and the absence of infectious organisms at bronchoalveolar lavage (BAL) or lung biopsy.6 Histopathologic findings associated with IPS include diffuse alveolar damage with hyaline membranes, accompanied by bronchiolar inflammation with lymphocyte infiltrates.52
Two specific histopathological patterns have been found in murine models: dense mononuclear cell infiltrates around pulmonary vessels and bronchioles and an acute pneumonitis involving interstitium and alveolar spaces.53 The lung is susceptible to immune-mediated injury by T-lymphocyte activation and inflammatory cytokine secretion.54 Specifically, TNF-α contributes to EC injury and apoptosis and directs leukocyte recruitment by regulating pulmonary chemokine expression.13,53,55
The definition of IPS encompasses various forms of pulmonary injury, depending on the main lung structure affected, including DAH and PERDS.
DAH generally develops immediately after HSCT and is characterized by progressive shortness of breath, cough, and hypoxemia, with or without fever.56 Diagnosis traditionally relies upon bloodier aliquots of BAL fluid, but frank hemoptysis is rare.57 Hemorrhagic pulmonary edema, due to diffuse alveolar damage with alveolar hemorrhage, is a characteristic pathologic finding. Despite prompt steroid therapy, the mortality from DAH is very high (70%-80%).56,57
PERDS typically occurs within 5 days of engraftment, after autologous or allogeneic HSCT. It is characterized by fever, dyspnea, and hypoxemia.58 Even when requiring mechanical support, PERDS after autologous HSCT or nonmyeloablative in vivo T-cell–depleted allogeneic HSCT typically responds promptly to corticosteroids and is associated with a favorable prognosis.59-61
Lung endothelial injury syndrome is probably the entity for which the literature is most confusing. By definition, the diagnosis must be made after exclusion of an infectious etiology. Older literature most likely included nondiagnosed viral infections. In a recent study from the Fred Hutchinson Cancer Center group in Seattle, WA, 69 BAL samples from HSCT patients diagnosed with IPS were retested for 28 viral, bacterial, and fungal pathogens using polymerase chain reaction. An occult infectious etiology was found in almost 60% of cases.62
Furthermore, DAH should be considered a pathological finding rather than a syndrome, because it could be linked to an undiagnosed infection or drug toxicity. Finally, PERDS in the allogeneic setting may be challenging to differentiate from CLS or hyperacute GVHD.
CLS
CLS is a systemic endothelial disorder that is pathogenically related to the capillary endothelium injury, because of cytokines such as vascular endothelial growth factor (VEGF).63 It is characterized clinically by a loss of intravascular fluids into the interstitial space due to a diffusely increased capillary permeability. In its most extreme form, it can lead to a profound hemodynamic collapse and multiorgan system failure.64 This syndrome is characterized by the development, in the first 15 days after HSCT, of weight gain (>3% within 24 hours) and a generalized edematous syndrome (ascites, pleural effusion, or pericarditis) that typically do not respond to furosemide treatment.
Other features that are occasionally observed include renal insufficiency, tachycardia, hypotension, and hypoalbuminemia. Adequate estimation of the incidence of CLS is difficult because there are no well-established clinical criteria. Moreover, distinction from other EC activation syndromes can be complicated.
Identified risk factors are granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor use, high cumulative doses of chemotherapy in the pre-HSCT phase, and unrelated or HLA-mismatched donor graft (and a concomitant GVHD).64-66
Engraftment syndrome
ES, formerly known as autoaggression syndrome, or aseptic shock syndrome involves a massive release of proinflammatory cytokines (IL-2, TNF-α, IFN-γ, IL-6), M-GSF, EPO, products of degranulation and oxidative metabolism of neutrophils, and systemic endothelial damage. In the context of allogeneic HSCT, ES has been principally seen after nonmyeloablative and cord blood HSCT.67,68, It is also described after autologous HSCT at the time of neutrophil engraftment.69,70 During neutrophil recovery following cytotoxic chemotherapy or stem cell transplantation, a spiraling production and release of cytokines and products of neutrophil degranulation and oxidative metabolism occur.71 These can lead to local (eg, in the lung where the neutrophils may initially be sequestered) and systemic (eg, fever and a diffuse increase in capillary permeability) tissue injury.3,72
Spitzer defined major and minor criteria for ES diagnosis.4 The 3 major criteria are noninfectious fever, erythroderma involving ≥25% body surface area not attributed to medication, and noncardiogenic pulmonary edema. The 4 minor criteria are total bilirubin ≥2 mg/dL or transaminases at least twice the normal levels, serum creatinine ≥2 times greater than baseline, weight gain ≥2.5% of baseline body weight, or unexplained encephalopathy. Essential for the diagnosis of ES are 3 major criteria or 2 major criteria plus 1 minor criterion.
In a recent retrospective single-center study of 927 consecutive allogeneic HSCT recipients, 13% developed ES. These patients had higher incidences of acute GVHD and nonrelapse mortality and poorer overall survival than patients not developing ES. Plasma levels of suppression of tumorigenicity 2, IL-2 receptor α, and TNF receptor 1 were significantly elevated in ES patients.73
For patients with progressive or symptomatic postallogeneic ES, particularly in cases of pulmonary involvement, corticosteroids can be useful.74-76
Lee et al described a “preengraftment syndrome” that shares clinical features with ES, even though it appears earlier, in the preengraftment period.77
Principles of therapy
Several therapeutic approaches have been proposed for some of these syndromes, but the mortality remains unacceptably high. Supportive care measures and high-dose IV corticosteroids are essential to managing most of these forms. More recently, a deeper understanding of the pathogenic mechanisms underlying these syndromes is driving the development of novel and, possibly, more effective treatments. Table 2 summarizes principal prospective studies.
Principal studies reporting patients treated for endothelial syndromes
| References | Characteristics | Patients, n | Median age (range), y | Diagnostic criteria | Onset, days from HSCT (range) | Response rate or survival, n (%) |
|---|---|---|---|---|---|---|
| TA-TMA | ||||||
| Rituximab | ||||||
| Au et al93 | Retrospective rituximab (375 mg/m2 per wk × 4) | 5 | 48 (1-56) | BMT-CTN | 42 (18-505) | CR 4/5 (80) |
| Jodele et al44 | Prospective biological study; rituximab (375 mg/m2 per wk for 2-10 courses) | 6 | 3.5 (2-17) | O-TMA | 32 (9-111) | CR 4/6 (67) |
| Defibrotide | ||||||
| Corti et al94 | Retrospective (40 mg/kg p.o. daily for 30-50 d) | 12 | 26 (1-55) | Hemolytic anemia, antiglobulin test (−), schystocytosis, haptoglobin ↓, LDH ↑, thrombocytopenia, CNS or renal involvement | 47 (9-100) | CR 5/12 (42)/PR 3/12 (25) |
| Yeates et al95 | Retrospective: median dose in ped: 30 mg/kg per d (range, 25-60); in ad: 400 mg/kg per d (range, 200-800) | 39 (22 ped/17 ad) | 8 (0-17)/40 (18-63) | O-TMA | 51 (10-215)/33 (15-152) | 17/22 (77)/13/17 (77) |
| Vincristine | ||||||
| Silva et al96 | Retrospective: 1-2 mg IV weekly or 0.2 mg IV daily (associated with PE and CCS) | 6 | 24.5 (13-35) | Hemolytic anemia, antiglobulin test (−), LDH ↑, thrombocytopenia, CNS or renal involvement | 49 (19-110) | CR 1/6 (17), PR 2/6 (33) |
| Mateos et al97 | Retrospective: 1 mg once daily IV on days 1, 4, and 8 ± day 11 | 7 | 18 (6-48) | Anemia, LDH ↑, thrombocytopenia, schistocytosis | 49 (25-276) | CR 6/7 (86) |
| Eculizumab | ||||||
| Jodele et al49 | Prospective: induction dose: 600 (<40 kg) or 900 mg (>40 kg); maintenance dose: 900 or 1200 every 14 d | 6 | 5 (2-10) | O-TMA | 49 (6-390) | CR 4/6 (67) |
| de Fontbrune et al48 | Retrospective: induction dose: 900 mg weekly for 4 wk, followed by maintenance (1200 mg every 14 d) | 12 (3 ped/9 ad) | 39 (1.2-66) | + | 121 (37-455) | OR 6/12 (50) |
| Jodele et al78 | Prospective: induction dose: 600 mg (<40 kg) or 900 mg (>40 kg), subsequent doses based on CH50 monitoring | 18 | 4.6 (2-15.2) | Refined TMA | 30 (18-55) | CR 11/18 (61) |
| Bohl et al98 | Retrospective: 900 mg once weekly until clinical response, followed by maintenance therapy (1200 mg every 14 d) | 24 | 48 (23-66) | O-TMA | 180 (15-984) | OR 13/15 (93) |
| IPS | ||||||
| Anti-TNF | ||||||
| Yanik et al80 | Retrospective anti-TNF+CCS (2 mg/kg per d) | 15 | 21 (1-60) | 14 (9-87) | 10/15 (66) | |
| Yanik et al81 | Prospective randomized ARM A: anti-TNF+CCS (2 mg/kg per d). ARM B: placebo + CCS (2 mg/kg per d). | A: 16; B: 18 | A: 10/16 (62); B: 12/18 (66.7) | |||
| Yanik et al92 | Prospective phase 2 anti-TNF+CCS (2 mg/kg per d) | 39 | 11 (1-17) | 20 (6-119) | 20/39 (71) | |
| Thompson et al82 | Retrospective anti-TNF+CCS (2 mg/kg per d) | 23 | 56 (35-72) | 233 (104-555) | 10/23 (43) |
| References | Characteristics | Patients, n | Median age (range), y | Diagnostic criteria | Onset, days from HSCT (range) | Response rate or survival, n (%) |
|---|---|---|---|---|---|---|
| TA-TMA | ||||||
| Rituximab | ||||||
| Au et al93 | Retrospective rituximab (375 mg/m2 per wk × 4) | 5 | 48 (1-56) | BMT-CTN | 42 (18-505) | CR 4/5 (80) |
| Jodele et al44 | Prospective biological study; rituximab (375 mg/m2 per wk for 2-10 courses) | 6 | 3.5 (2-17) | O-TMA | 32 (9-111) | CR 4/6 (67) |
| Defibrotide | ||||||
| Corti et al94 | Retrospective (40 mg/kg p.o. daily for 30-50 d) | 12 | 26 (1-55) | Hemolytic anemia, antiglobulin test (−), schystocytosis, haptoglobin ↓, LDH ↑, thrombocytopenia, CNS or renal involvement | 47 (9-100) | CR 5/12 (42)/PR 3/12 (25) |
| Yeates et al95 | Retrospective: median dose in ped: 30 mg/kg per d (range, 25-60); in ad: 400 mg/kg per d (range, 200-800) | 39 (22 ped/17 ad) | 8 (0-17)/40 (18-63) | O-TMA | 51 (10-215)/33 (15-152) | 17/22 (77)/13/17 (77) |
| Vincristine | ||||||
| Silva et al96 | Retrospective: 1-2 mg IV weekly or 0.2 mg IV daily (associated with PE and CCS) | 6 | 24.5 (13-35) | Hemolytic anemia, antiglobulin test (−), LDH ↑, thrombocytopenia, CNS or renal involvement | 49 (19-110) | CR 1/6 (17), PR 2/6 (33) |
| Mateos et al97 | Retrospective: 1 mg once daily IV on days 1, 4, and 8 ± day 11 | 7 | 18 (6-48) | Anemia, LDH ↑, thrombocytopenia, schistocytosis | 49 (25-276) | CR 6/7 (86) |
| Eculizumab | ||||||
| Jodele et al49 | Prospective: induction dose: 600 (<40 kg) or 900 mg (>40 kg); maintenance dose: 900 or 1200 every 14 d | 6 | 5 (2-10) | O-TMA | 49 (6-390) | CR 4/6 (67) |
| de Fontbrune et al48 | Retrospective: induction dose: 900 mg weekly for 4 wk, followed by maintenance (1200 mg every 14 d) | 12 (3 ped/9 ad) | 39 (1.2-66) | + | 121 (37-455) | OR 6/12 (50) |
| Jodele et al78 | Prospective: induction dose: 600 mg (<40 kg) or 900 mg (>40 kg), subsequent doses based on CH50 monitoring | 18 | 4.6 (2-15.2) | Refined TMA | 30 (18-55) | CR 11/18 (61) |
| Bohl et al98 | Retrospective: 900 mg once weekly until clinical response, followed by maintenance therapy (1200 mg every 14 d) | 24 | 48 (23-66) | O-TMA | 180 (15-984) | OR 13/15 (93) |
| IPS | ||||||
| Anti-TNF | ||||||
| Yanik et al80 | Retrospective anti-TNF+CCS (2 mg/kg per d) | 15 | 21 (1-60) | 14 (9-87) | 10/15 (66) | |
| Yanik et al81 | Prospective randomized ARM A: anti-TNF+CCS (2 mg/kg per d). ARM B: placebo + CCS (2 mg/kg per d). | A: 16; B: 18 | A: 10/16 (62); B: 12/18 (66.7) | |||
| Yanik et al92 | Prospective phase 2 anti-TNF+CCS (2 mg/kg per d) | 39 | 11 (1-17) | 20 (6-119) | 20/39 (71) | |
| Thompson et al82 | Retrospective anti-TNF+CCS (2 mg/kg per d) | 23 | 56 (35-72) | 233 (104-555) | 10/23 (43) |
Note the lack of studies concerning endothelial syndromes other than TA-TMA and IPS.
ad, adults; BMT-CTN, Blood and Marrow Transplant Clinical Trials Network criteria; CCS, corticosteroid; CH50, complement inhibition 50; CR, complete response; LDH, lactate dehydrogenase; OR, overall response; O-TMA, overall TMA criteria; PE, plasma exchange; ped, pediatrics; p.o., by mouth; PR, partial response.
Corticosteroids
Although scientific evidence is limited in this setting, steroids remain the most used agent in these conditions, primarily for their anti-inflammatory properties, which might prevent further endothelial damage.
In the specific context of ES, corticosteroids (usually methyl-prednisolone, 1-2 mg/kg per day for 3 days, with subsequent tapering over 1 week) represent the optimal strategy.74-76 Because of the difficulty with excluding the infectious origin of the fever, the need to confirm that the fever does not respond to empirical antibiotic therapy and that cultures are negative is mandatory. A complete resolution of symptoms is achieved in 1 to 5 days in >80% of cases; however, in some cases, the symptoms reappear after stopping steroid therapy.74,75
Corticosteroids were also used for the treatment of CLS, even if clinical results were far from satisfying, because the response rate was usually poor.
Anticomplement agents
Because of the complement-based pathophysiology, anticomplement agents have a role in the setting of TA-TMA. Eculizumab is a monoclonal antibody directed against complement fraction 5 (C5) and blocks the terminal pathway of the complement cascade and membrane attack complex formation. After a first index case,47 the French Society of Blood and Bone Marrow Transplantation and Cellular Therapy reported a series of 12 TA-TMA patients treated with eculizumab, with a hematological response in 50% of cases and 33% overall survival.48 These data are in agreement with the pediatric experience of 6 patients in Cincinnati, which initially showed a 66% response rate.49
A more recent study, from the same group, on a cohort of 18 patients reported a 61% response rate, with 56% overall survival, as compared to historical control (9%).78 In this study, complement activation (based on C5b-C9 plasma levels) correlated with faster eculizumab clearance, which may result in subtherapeutic plasma levels of eculizumab (measured as hemolytic complement activity) and lack of a clinical response. Eculizumab should be considered an effective treatment option for patients developing TA-TMA; nevertheless, its use in the setting of TA-TMA remains off-label.
A second generation of complement modulators is in preclinical or clinical development in the setting of paroxysmal nocturnal hemoglobinuria and other hemolytic complement-based diseases. Clinical trials in patients with TA-TMA are lacking. Novartis designed a randomized study to analyze the efficacy and safety of LFG316 (NCT02763644), another anti-C5 compound, but the trial was closed prematurely because of the small number of inclusions.
Anti–TNF-α agents
Based on experimental studies, anti–TNF-α agents have been suggested as a treatment strategy for IPS.54,56,79 Yanik et al proposed etanercept (subcutaneously, at a dose of 0.4 mg/kg twice weekly for a maximum of 8 doses) in combination with systemic steroids and antimicrobial therapy in 15 patients with IPS.80 Ten of the 15 patients had a complete response (defined as the ability to discontinue supplemental oxygen support during study therapy), and survival at day 28 from the first etanercept dose was 76%. Results were also reported from a larger phase 2 study on the use of systemic corticosteroids (2 mg/kg per day) plus etanercept (0.4 mg/kg twice weekly, 8 doses) in 39 pediatric patients (median age, 11 years; range, 1-17). Complete response rate was 71%, with a median time to response of 10 days (range, 1-24). However, results of a randomized double-blind placebo-controlled trial conducted by the same group comparing etanercept plus steroid therapy vs placebo plus steroids showed no difference between the 2 treatment arms.81 Thompson et al published a retrospective study of 23 adult patients who developed posttransplant IPS treated by steroids and etanercept using a homogeneous treatment schedule.82 Complete response rate was 43%, with a 2-year overall survival of 67% among responding patients.
CNI withdrawal
CNI discontinuation, especially in cases of TA-TMA, is a common practice in most transplant centers and is an object of general recommendations.32 However, this approach is not supported by robust evidence, and it may be deleterious early post-HSCT, especially in cases of underlying GVHD.
In a large retrospective series aiming to analyze the risk factors and outcomes of TA-TMA, the effect of cyclosporine withdrawal was not confirmed in the multivariable analysis.83 Very often, these medications are stopped in patients showing clinical features of TA-TMA, VOD/SOS, ES, or CLS; however, because of a lack of prospective data, we are far from being able to recommend or caution against this action.
Additionally, many anti-infectious agents or conditions could contribute to an increased risk for organ toxicity in these settings. Given the lack of conclusive data, we recommend that all adjunctive approaches, including managing GVHD prophylaxis and treatment, should be evaluated on a case-by-case basis in transplant recipients presenting with EC activation syndromes and that the benefits and risks be considered carefully for each patient.
Other treatments
Therapeutic plasma exchange (PE) has been shown to be effective in TTP (response rate, 78%-91%) via the removal of anti–ADAMTS-13 autoantibodies, whereas its response rate is lower in TA-TMA (27%-80%).84,85 Based on previous reports that showed poor outcomes after upfront PE for TA-TMA, the Blood and Marrow Transplant Clinical Trials Network committee recommended against its routine use.32 However, recent data have revealed improved outcomes, with response rates > 50% when PE is initiated early in the disease course.86 Nevertheless, as mentioned above, more recent trials with eculizumab have shown higher response rates.49,78
The treatment of endothelial complications is often empiric, because clinical evidence is lacking. Intravenous immunoglobulins87 and human anti-VEGF antibody (bevacizumab)88 have been used successfully in the setting of CLS; however, these data remain anecdotal and should be confirmed in larger series.
The treatment of endothelial lung complications is even more disappointing, because, in most cases, lung involvement is the last irreversible event of systemic endothelial damage. Although there is no treatment for frank DAH, veno-venous hemofiltration may be considered for IPS.89 In this setting, prompt initiation of the procedure is crucial, eventually leading to a survival benefit in some patients. Steroid use has not demonstrated an advantage, and no agent or combination of agents has been proven superior.
Prospective studies addressing the treatment of other forms of endothelial activation syndromes are lacking. Irrespective of the type of endothelial activation syndrome, the role of supportive care is crucial.
Controversial issues and management strategies
Despite significant advances in critical care medicine, EC activation syndromes remain frequently fatal complications following allogeneic HSCT. All of these endothelial injury syndromes may overlap with acute GVHD and, sometimes, the clinical distinction among these forms and GVHD can be challenging. A common origin shared with acute GVHD had already been proposed some years ago by Tichelli and Gratwohl.14 They suggested that these syndromes should be regarded as “endothelial” forms of acute GVHD, supporting the hypothesis of vascular endothelium as a target of GVHD. The same investigators consider that atherosclerosis and late cardiovascular events belong to the same family of endothelial disorders.14,19
We want to share the idea that postallogeneic HSCT endothelial syndromes represent a complex range of diseases, in which the predominant manifestation depends on the type of organ-related injury and overlap with GVHD (Figure 1). The lack or incompleteness of reliable preclinical models and large ex vivo studies in humans does not allow for a full understanding of all pathophysiological mechanisms. This represents a further limitation to the development of novel target treatments that may improve the dismal prognosis of patients.
To improve agreement on a better categorization of these syndromes, we strongly suggest excluding VOD/SOS as a purely endothelial disorder, because other mechanisms play a role in its pathogenesis. Indeed, although the endothelial damage, in response to several pathogenic noxa, represents the first event and is at the origin of (1) denudation of the sinusoidal lining, (2) extravasation of red cells into the space of Disse, (3) embolization of blood stream and obstruction of sinusoidal flow, and (4) consequent hemodynamic changes with portal hypertension,10 hepatocyte dysfunction also contributes directly to VOD/SOS pathophysiology. An impaired function of the glutathione enzymatic system in hepatocytes was described in murine models of VOD/SOS with subsequent alteration of pharmacokinetics of several drugs (eg, cyclophosphamide and busulfan).90,91 Previous liver disease or previous exposure to hepatotoxic drugs impairs this system, leading to the accumulation of toxic metabolites that may injure sinusoidal ECs, as well as hepatocytes.91
Based on the existing evidence discussed in previous paragraphs, as well as on experience from Saint Louis Hospital, we want to propose a classification and treatment algorithm for EC activation syndromes (Figure 2) that takes into account the critical limitations imposed by the absence of clinical trials.
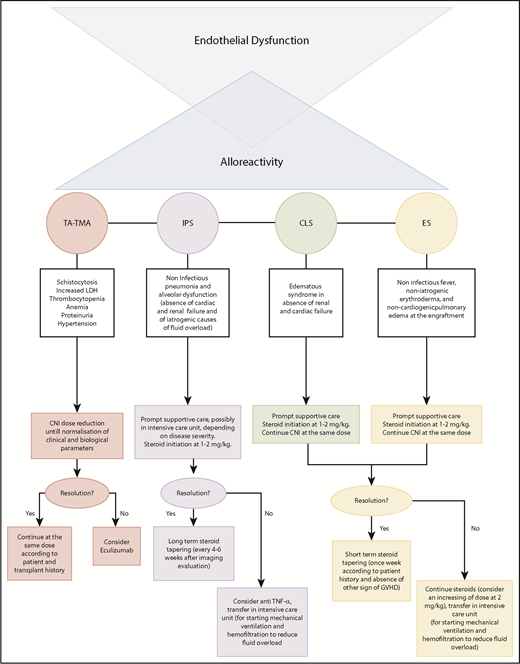
Particularly, in TA-TMA, we recommend using eculizumab in patients not responding to reduced doses of CNIs. We propose adopting the classic dosing schedule used in HUS (induction dose of 900 mg eculizumab IV every week for 4 weeks, followed by a maintenance dose of 1200 mg every 14 days), as retrospectively and prospectively assessed.48,49 Treatment should be continued until hematological response, defined as the normalization of lactate dehydrogenase and haptoglobin, disappearance of schistocytes, and transfusion independence. The discontinuation of CNIs, as well as the introduction of steroids solely as GVHD prophylaxis, should be evaluated carefully.
Although strong evidence is lacking, the use of steroids in IPS, together with prompt supportive care, should be considered a possible frontline strategy. Doses may vary between 1 and 2 mg/kg according to disease severity and time from transplant. In cases of resolution, we recommend a long-term tapering of steroid treatment, with close clinical and radiological monitoring. Anti–TNF-α agents can be used as adjuvant treatment in subjects who do not respond to corticosteroids. Based on the results of 2 prospective trials,81,92 we recommend using etanercept at a dose of 0.4 mg/kg twice weekly (maximum 25 mg per dose) for 1 month in adult and pediatric patients. We also recommend steroids, at a dose of 1-2 mg/kg without CNI withdrawal, in subjects with CLS or ES.
Prompt supportive care is essential for prevention of multiorgan failure in severe forms. Antagonism of the VEGF pathway, exploration of the complement pathway in EC activation syndromes other than TA-TMA, and the study of prophylactic drugs for endothelial dysfunction in pretransplant settings can represent other directions for future investigations.
In conclusion, accurate diagnoses and risk assessments, through a reconsideration of precise classification and definitions, are urgently needed to improve outcomes and survival for patients with EC activation syndromes. Unfortunately, prospective clinical trials are lacking, and as a result of the low incidence of these disorders, research on new therapeutic targets continues to be challenging.
Authorship
Contribution: S.P. and G.S. conceived the review and wrote the manuscript; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Gérard Socie, Hematology Transplantation, Saint Louis Hospital, Ave Claude Vellefaux, 75010 Paris, France; e-mail: gerard.socie@aphp.fr.

