

Key Points
CMV reactivation was associated with the maturation of reconstituting NK cells from BM, but not PB, unrelated donor grafts.
CMV reactivation was associated with CD8+, but not CD4+, T-cell recovery, more so after BM than PB unrelated donor grafts.
Introduction
Delayed immune reconstitution after allogeneic hematopoietic cell transplantation (HCT) increases the risk of relapse,1 infection,2 and secondary malignancy.3 Natural killer (NK) cells, the earliest reconstituting immune cells, reach normal levels within weeks after HCT4,5 and, with T cells, contribute to the graft-versus-tumor effect.6 Understanding NK cell reconstitution can help predict risks of relapse and identify novel therapeutic interventions.
Cytomegalovirus (CMV) exposure results in expansion of a unique subset of NK cells (termed adaptive) that exhibit properties of immune memory.7 This terminally differentiated CD56dimNKG2C+CD57+ NK-cell subset has potent antibody-dependent cytolytic function and persistence long after resolution of infection.7-9 Adaptive NK cells have been associated with protection against leukemia relapse10-12 and improved disease-free survival13 after HCT.
We asked whether the effect of CMV reactivation on NK- and T-cell reconstitution after HCT is influenced by graft source. Using unique samples from a large randomized study of peripheral blood (PB) vs bone marrow (BM) unrelated donor HCT, we demonstrate that CMV reactivation is associated with more mature NK cell reconstitution after HCT with BM, but not PB grafts.
Methods
Clinical data and PB mononuclear cell samples were provided by the Blood and Marrow Clinical Trials Network (BMT CTN). BMT CTN‐0201 was a phase 3 randomized multicenter trial (2004-2009, 551 patients) comparing unrelated donor BM vs PB HCTs, with 2‐year survival as the primary end point.14 The trial demonstrated similar rates of acute graft-versus-host disease (aGVHD) but higher rates of chronic GVHD among PB graft recipients. An analysis of infections15 showed the BM group with a higher 2-year cumulative incidence of all infections and bacterial infections but similar 2-year cumulative incidence of CMV and fungal infections.
We included all patients with a day 100 PB mononuclear cell sample without relapse by day 100 (n = 259) (Table 1). We classified patients into 3 CMV groups: (1) patients who reactivated CMV by day 100 (CMVr), (2) CMV-seropositive patients who did not reactivate CMV by day 100 (CMV+), and (3) CMV-seronegative patients who did not reactivate CMV (CMV−). We compared the frequency of day 100 immune cell subsets among these 3 CMV groups.
Patient- and transplant-related characteristics
| Variable | Overall (n = 259) | BM (n = 126) | PB (n = 133) | P |
|---|---|---|---|---|
| Sex | 0.31 | |||
| Male | 152 (59) | 78 (62) | 74 (56) | |
| Female | 107 (41) | 48 (38) | 59 (44) | |
| Age at transplantation, y | 0.90 | |||
| Median (range) | 44 (0.4-67) | 42 (5-67) | 41 (0.4-67) | |
| Race | 0.20 | |||
| Nonwhite | 20 (8) | 7 (6) | 13 (10) | |
| White | 239 (92) | 119 (94) | 120 (90) | |
| CMV group | 0.11 | |||
| CMVr | 34 (13) | 22 (18) | 12 (9) | |
| CMV+ | 110 (43) | 53 (42) | 57 (43) | |
| CMV− | 115 (44) | 51 (41) | 64 (48) | |
| Disease group | 0.43 | |||
| Acute myeloid leukemia | 127 (49) | 59 (47) | 68 (51) | |
| Acute lymphoblastic leukemia | 59 (23) | 30 (24) | 29 (22) | |
| Chronic myeloid leukemia | 25 (10) | 13 (10) | 12 (9) | |
| Myelodysplastic syndrome | 40 (15) | 22 (18) | 18 (14) | |
| Chronic myelomonocytic leukemia | 4 (2) | 2 (2) | 2 (2) | |
| Myelofibrosis | 4 (2) | 0 (0) | 4 (3%) | |
| HLA match degree | 0.07 | |||
| 5/6 | 26 (10) | 17 (14) | 9 (7) | |
| 6/6 | 233 (90) | 109 (87) | 124 (93) | |
| Acute GVHD grade II to IV by day 100 | 0.52 | |||
| No | 143 (55) | 67 (53) | 76 (57) | |
| Yes | 116 (45) | 59 (47) | 57 (43) |
| Variable | Overall (n = 259) | BM (n = 126) | PB (n = 133) | P |
|---|---|---|---|---|
| Sex | 0.31 | |||
| Male | 152 (59) | 78 (62) | 74 (56) | |
| Female | 107 (41) | 48 (38) | 59 (44) | |
| Age at transplantation, y | 0.90 | |||
| Median (range) | 44 (0.4-67) | 42 (5-67) | 41 (0.4-67) | |
| Race | 0.20 | |||
| Nonwhite | 20 (8) | 7 (6) | 13 (10) | |
| White | 239 (92) | 119 (94) | 120 (90) | |
| CMV group | 0.11 | |||
| CMVr | 34 (13) | 22 (18) | 12 (9) | |
| CMV+ | 110 (43) | 53 (42) | 57 (43) | |
| CMV− | 115 (44) | 51 (41) | 64 (48) | |
| Disease group | 0.43 | |||
| Acute myeloid leukemia | 127 (49) | 59 (47) | 68 (51) | |
| Acute lymphoblastic leukemia | 59 (23) | 30 (24) | 29 (22) | |
| Chronic myeloid leukemia | 25 (10) | 13 (10) | 12 (9) | |
| Myelodysplastic syndrome | 40 (15) | 22 (18) | 18 (14) | |
| Chronic myelomonocytic leukemia | 4 (2) | 2 (2) | 2 (2) | |
| Myelofibrosis | 4 (2) | 0 (0) | 4 (3%) | |
| HLA match degree | 0.07 | |||
| 5/6 | 26 (10) | 17 (14) | 9 (7) | |
| 6/6 | 233 (90) | 109 (87) | 124 (93) | |
| Acute GVHD grade II to IV by day 100 | 0.52 | |||
| No | 143 (55) | 67 (53) | 76 (57) | |
| Yes | 116 (45) | 59 (47) | 57 (43) |
Data are presented as n (%), unless otherwise indicated.
CMVr, patients who reactivated CMV by day 100; CMV+, CMV-seropositive patients who did not reactivate CMV by day 100; CMV−, CMV-seronegative patients who did not reactivate CMV.
Fresh blood samples (without cryopreservation) were stained fresh within 30 hours of collection for analysis. The Translational Therapy Laboratory (Masonic Cancer Center, University of Minnesota) processed specimens, stained and acquired cells on an LSRII flow cytometer (BD Biosciences), and analyzed data using FlowJo (supplemental Methods). CD4+ and CD8+ T-cell subsets and CD56bright, CD56dim, and KIR+NKG2A− NK-cell subsets were enumerated. We used a cocktail of 3 KIR antibodies with the same fluorochrome (CD158a, CD158b, and NKB1) to measure the KIR+ population. We used KIR+NKG2A− expression on CD56dim NK cells to estimate the most mature NK-cell subset (as NKG2C expression was not available). NKG2C and NKG2A expression are generally mutually exclusive. Additionally, Malmberg et al showed that mature adaptive NK cells express increased KIR and low NKG2A.16 While the KIR+NKG2A− phenotype of CD56dim NK cells is an estimate, true adaptive NK cells would be proportional to this phenotype.
Statistical analysis
Patient- and treatment-related variables were compared between the treatment groups using a Student t test, χ2 test, or Fisher’s exact test as appropriate. The percentage of cell subsets was compared between groups using the Wilcoxon rank sum test. We used linear regression for each cell type by including graft source, CMV group, and their interaction as covariates. Because aGVHD can influence NK cell reconstitution,17 models adjusted or unadjusted for grade II to IV aGVHD by day 100 were compared to evaluate whether our results are influenced by aGVHD.
Results and discussion
In univariate analysis considering graft source as the only independent variable (supplemental Table 1), day 100 CD4+ T cells were significantly higher in the PB group (P < .0001), while total NK cells and their CD56bright subset were significantly higher in the BM group (P < .001).
The kinetics of lymphocyte reconstitution at 100 days is shown in Figure 1A-C. In both BM and PB subgroups, day 100 CD8+ T cells were higher in CMVr and CMV+ groups than the CMV− group (P < .001 in BM and P < .01 in PB). In both BM and PB subgroups, day 100 CD4+ T cells were reciprocally slightly lower in the CMVr group than the CMV+ and CMV− groups (not statistically significant). In regression modeling using an interaction term for graft source and CMV group, the interaction was significant for CD8+ T cells (P = .04), where the association of CMV reactivation with CD8+ T-cell recovery was more pronounced with BM. No significant interaction was found for CD4+ T cells (P = .53). These results suggest expansion of CD8+ T cells due to CMV reactivation (more prominently in BM recipients), consistent with potent immune imprinting of CMV on these cells.18,19 Specifically, CMV reactivation results in clonal expansion of effector memory CD8+ T cells with a compromised T-cell receptor repertoire, a linked global contraction of CD4+ and CD8+ naïve T cells,20 and possibly also a lower fraction of CMV-specific CD8+ T cells with robust cytokine response.21
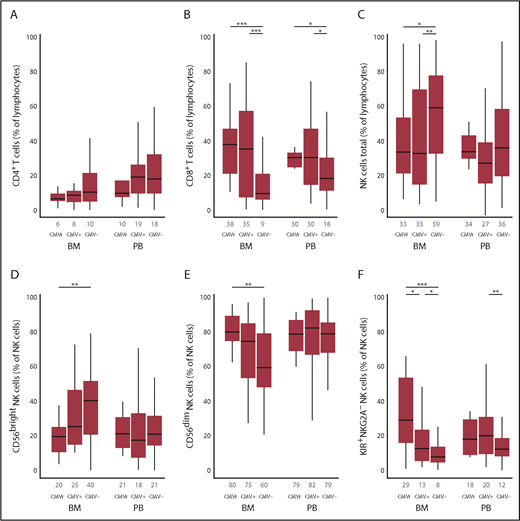
Association of CMV reactivation with T- and NK-cell reconstitution at 100 days in unrelated donor allografts using BM vs PB as a graft source. The frequency of CD4+ T cells (A), CD8+ T cells (B), and total NK cells (C) were determined based on a lymphocyte gate determined by forward and side scatter. The NK cells (CD56+/CD56− gate) were subsetted into CD56bright (D), CD56dim (E), and KIR+NKG2A− (F) NK cells in 3 groups based on CMV reactivation status and in BM vs PB groups separately. Blue numbers on the x-axis show medians. CMVr, patients who reactivated CMV by day 100; CMV+, CMV-seropositive patients who did not reactivate CMV by day 100; and CMV−, CMV-seronegative patients who did not reactivate CMV by day 100. *P < .05; **P < .01; ***P < .001.
Association of CMV reactivation with T- and NK-cell reconstitution at 100 days in unrelated donor allografts using BM vs PB as a graft source. The frequency of CD4+ T cells (A), CD8+ T cells (B), and total NK cells (C) were determined based on a lymphocyte gate determined by forward and side scatter. The NK cells (CD56+/CD56− gate) were subsetted into CD56bright (D), CD56dim (E), and KIR+NKG2A− (F) NK cells in 3 groups based on CMV reactivation status and in BM vs PB groups separately. Blue numbers on the x-axis show medians. CMVr, patients who reactivated CMV by day 100; CMV+, CMV-seropositive patients who did not reactivate CMV by day 100; and CMV−, CMV-seronegative patients who did not reactivate CMV by day 100. *P < .05; **P < .01; ***P < .001.
In the BM subgroup, CMV reactivation was associated with fewer total (P < .05) and CD56bright (P < .01) NK cells compared with the CMV− group (Figure 1C-D). However, more mature CD56dim NK cells expanded in the CMVr group than the CMV− group (P < .01; Figure 1E). The frequency of KIR+NKG2A− NK cells was highest in the CMVr group, followed by the CMV+ (P < .05) and CMV− groups (P < .05) (Figure 1F). The results for absolute counts (supplemental Figure 1) were consistent with our main findings.
These findings are consistent with previous reports of NK-cell maturation following CMV reactivation.9,11 In the PB subgroup, there was an increase in mature KIR+NKG2A− NK cells in the CMV+ group, but not the CMVr group, compared with the CMV− group (P < .01). In regression modeling using an interaction term for graft source and CMV group, the interaction was significant for KIR+NKG2A− NK cells, where the effect of CMV reactivation on KIR+NKG2A− NK-cell recovery was more pronounced with BM (P = .03). No significant graft source interaction was found for total (P = .48), CD56bright (P = .30), or CD56dim NK cells (P = .30). CMV group was not associated with relapse or nonrelapse mortality in either the BM or PB group. Since the results for the graft source × CMV group interaction in regression models adjusted or unadjusted for aGVHD were very similar, grade II to IV aGVHD by day 100 did not confound our main findings (supplemental Table 2).
The association of CMV reactivation with NK and other lymphoid cell immune reconstitution depends on graft source, with a stronger association in BM recipients compared with PB. This association is CMV dose dependent in recipients of BM, but not PB grafts, where detectable CMV reactivation had the greatest association compared with latent CMV persistence in CMV-seropositive patients without detectable reactivation. Why the association of CMV reactivation with NK-cell maturation is graft-source dependent may have several explanations. Since the CMV reservoir is thought to occur in myeloid cells, this latent pool may differ between BM and PB grafts either directly from the graft source or perhaps acting through host cells. Graft source compositional differences in T cells (frequency, differentiation status, and CMV specificity), dendritic cells, and their subsets may also contribute.22 It is also plausible that BM is the source for certain subsets of NK cells. Our findings could suggest that a CMV-responsive precursor may be enriched in BM harvests and less so in granulocyte-colony stimulating factor–mobilized PB collections. Tissue-resident lymphocytes and their precursors may differ between graft sources. While it is believed that CD56bright NK cells develop into CD56dim NK cells with maturation that is linear and stage specific,23 this remains uncertain and has not been well studied in cells from BM vs PB. Nonhuman primate data suggest parallel lineages rather than sequential.24 Other possibilities include blunting of the CMV response with a Th2-biased effect induced by granulocyte-colony stimulating factor mobilization in PB grafts25 and higher levels of homeostatic cytokines in BM recipients as a key driver of the immune effects. Lastly, it is possible that the immune reconstitution associations observed are indirect and attributable to a higher incidence of CMV reactivation by day 100 (18% in BM and 9% in PB patients in our analysis, P = .04) as the dominant driver of immune differences.
The analysis performed here was on fresh samples obtained from the clinical trial, and higher resolution NK immunophenotypic panels were not available at the time of the initial trial design. Nonetheless, this is the first report on CMV-induced NK-cell immune reconstitution that differs between BM and PB graft sources. Further studies are needed to understand the mechanism and manipulate it for therapeutic purposes.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the BMT CTN for providing the samples.
Research reported in this publication was supported by the National Institutes of Health, National Cancer Institute (grant P30CA077598) utilizing the Biostatistics and Bioinformatics Core and the Biospecimen Repository in the Translational Therapy Laboratory Shared Resource of the Masonic Cancer Center, University of Minnesota and by the National Center for Advancing Translational Sciences of the National Institutes of Health (award number UL1-TR002494). National Institutes of Health, National Cancer Institute grants P01 CA111412 and P01 CA65493 provided partial support. BMT CTN is supported by the National Institutes of Health, National Heart, Lung, and Blood Institute and National Cancer Institute (grant U10HL069294). The Department of the Navy, Office of Navy Research, and the National Marrow Donor Program also provided support for the BMT CTN-0201 study. Enrollment support was provided by DKMS (Deutsche KnochenMarkSpenderdatei) Germany.
Any views, opinions, findings, conclusions or recommendations expressed in this material are those of the authors and do not reflect the views of the official policy or position of the above-mentioned parties.
Authorship
Contribution: A.R., S.C., D.J.W., and J.S.M. designed the study and critically evaluated the results; X.L. analyzed the data; A.R., D.J.W., and J.S.M. wrote the manuscript; and S.C., C.A., E.K.W., C.G.B., and F.C. critically reviewed and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jeffrey S. Miller, Division of Hematology, Oncology, and Transplantation, Department of Medicine, University of Minnesota, 420 Delaware St SE, MMC 480, Minneapolis, MN 55455; e-mail: mille011@umn.edu.

