

Key Points
Myeloma patients progressing on BCMA-targeted therapy can maintain BCMA expression and still respond to different BCMA-targeted therapy.
These observations suggest this patient population could be included in ongoing BCMA-targeted therapy trials.
Introduction
B-cell maturation antigen (BCMA), a cell surface receptor expressed on myeloma cells,1,2 is emerging as a promising target for myeloma therapy. Several novel BCMA-specific therapies, including antibody-drug conjugates, chimeric antigen receptor (CAR) T cells, and bispecific antibodies, have demonstrated significant activity in early-phase trials in relapsed/refractory myeloma.3-6 Although transient downregulation or loss of BCMA expression has been described following BCMA CAR T-cell therapy, in most cases BCMA expression is maintained at progression,4,6 suggesting patients may be able to respond to a subsequent BCMA-targeted modality. As a parallel example, acute lymphoblastic leukemia patients relapsing after the CD19-bispecific antibody blinatumomab can respond to CD19-specific CAR T cells.7 The promising results observed in initial studies make it likely that multiple BCMA-targeted therapeutics with different mechanisms of action will eventually be commercially available. However, most current trials of BCMA-targeted therapies exclude patients previously treated with other BCMA-targeted agents. The absence of clinical trial data on patients sequentially exposed to these agents will pose a challenge to myeloma patients and their physicians, who can choose among multiple available BCMA-targeted therapies. Here, we describe 2 patients who progressed after 1 BCMA-targeted therapy and then responded to a subsequent BCMA-targeted therapy. These cases demonstrate that BCMA-targeted therapies may be beneficial in patients previously exposed to other BCMA-targeted agents and suggest that this population should be included in future trials.
Case descriptions
Patient 1
Patient 1 is a 59-year-old woman with immunoglobulin G (IgG) λ myeloma who was treated on a phase 1 trial of BCMA CAR T cells (CD3ζ/4-1BB domains, lentiviral vector, University of Pennsylvania/Novartis study)6 in June 2016. At enrollment, she was refractory to bortezomib, lenalidomide, carfilzomib, pomalidomide, and daratumumab, with 10 prior lines of therapy. She had 90% plasma cells in the marrow, with gain 1q, t(4;14), and deletion 17p by fluorescence in situ hybridization. She received 5 × 108 CAR T cells without lymphodepleting chemotherapy, achieving a minimal response (MR; Figure 1A) with reduction of marrow plasma cells to 20% at day 28. By month 2, she was starting to progress with isolated rise in serum free light chains, confirmed at month 3. Serial assessment of BCMA expression on her myeloma cells by flow cytometry (as previously described)6 showed a modest decrease in BCMA staining intensity at day 28 compared with baseline (mean fluorescence intensity, 1844 to 832; Figure 1B), but continued expression of BCMA on the majority of her myeloma cells. She subsequently enrolled on a phase 1 trial of the antibody-drug conjugate (ADC) GSK2857916 (belantamab mafodotin), an anti-BCMA monoclonal antibody conjugated to the microtubule inhibitor MMAF.3 She received 2 doses at 3.4 mg/kg IV every 3 weeks, again achieving an MR by serum M-spike but with more substantial reduction in serum free λ light chains (Figure 1A), as well as a decrease in bone marrow plasma cells from 38% pretreatment to <5% on day 125. Therapy was held for 6 weeks because of corneal toxicity, and then restarted at 2.55 mg/kg for 2 additional doses. Serum M-spike continued to decrease but she again had light chain progression and came off therapy in January 2017. A February 2017 biopsy of a focal bone lesion demonstrated continued BCMA expression on myeloma cells by an immunohistochemistry assay8 (Figure 1C). She subsequently received several additional therapies, including salvage autologous stem cell transplant, but ultimately died in September 2018.
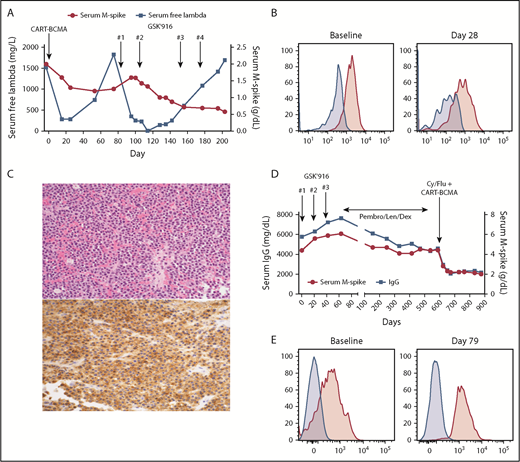
Treatment course and BCMA expression for patients 1 and 2. (A) Serum M-spike and free λ light chain levels for patient 1 are depicted over time, with treatment timepoints depicted by arrows. (B) Bone marrow aspirate cells from patient 1 pretreatment and 28 days post-BCMA CAR T-cell infusion were gated on live myeloma cells (CD45−CD38+CD19−CD56+λ+) and assessed for BCMA expression (pink histogram) as described in Cohen et al.6 Fluorescence minus 1 control (blue histogram) is also shown. (C) Biopsy of a focal bone lesion at progression post-GSK2857916 in patient 1 showed sheets of plasma cells (hematoxylin and eosin; top), with persistent BCMA expression by immunohistochemistry (bottom).8 Magnification ×200. (D) Serum M-spike and IgG levels for patient 2 are depicted over time, with treatment timepoints depicted by arrows. (E) Bone marrow aspirate mononuclear cells from patient 2 pretreatment and 79 days post-BCMA CAR T-cell infusion was assessed for BCMA expression by flow cytometry, gating on live myeloma cells (CD45−CD38+CD138+CD19−, pink histograms) or nonplasma cells (CD45+CD38−CD138−, blue histograms). Patient 1 treated on NCT025461676 ; patient 2 treated on NCT03288493.9 Cy, cyclophosphamide; Dex, dexamethasone; Flu, fludarabine; GSK′916, GSK285916 (NCT02064387)3 ; Len, lenalidomide; Pembro, pembrolizumab; qPCR, quantitative polymerase chain reaction.
Treatment course and BCMA expression for patients 1 and 2. (A) Serum M-spike and free λ light chain levels for patient 1 are depicted over time, with treatment timepoints depicted by arrows. (B) Bone marrow aspirate cells from patient 1 pretreatment and 28 days post-BCMA CAR T-cell infusion were gated on live myeloma cells (CD45−CD38+CD19−CD56+λ+) and assessed for BCMA expression (pink histogram) as described in Cohen et al.6 Fluorescence minus 1 control (blue histogram) is also shown. (C) Biopsy of a focal bone lesion at progression post-GSK2857916 in patient 1 showed sheets of plasma cells (hematoxylin and eosin; top), with persistent BCMA expression by immunohistochemistry (bottom).8 Magnification ×200. (D) Serum M-spike and IgG levels for patient 2 are depicted over time, with treatment timepoints depicted by arrows. (E) Bone marrow aspirate mononuclear cells from patient 2 pretreatment and 79 days post-BCMA CAR T-cell infusion was assessed for BCMA expression by flow cytometry, gating on live myeloma cells (CD45−CD38+CD138+CD19−, pink histograms) or nonplasma cells (CD45+CD38−CD138−, blue histograms). Patient 1 treated on NCT025461676 ; patient 2 treated on NCT03288493.9 Cy, cyclophosphamide; Dex, dexamethasone; Flu, fludarabine; GSK′916, GSK285916 (NCT02064387)3 ; Len, lenalidomide; Pembro, pembrolizumab; qPCR, quantitative polymerase chain reaction.
Patient 2
Patient 2 is a 49-year-old man with IgG κ myeloma who was treated on the GSK2857916 phase 1 trial starting in August 2016. At enrollment, he had received 5 prior lines and was refractory to carfilzomib, lenalidomide, daratumumab, elotuzumab, and pomalidomide. Baseline bone marrow had 70% plasma cells with normal cytogenetics. He received 3 doses of GSK2857916 at 3.4 mg/kg every 3 weeks, with continued progression on therapy (Figure 1D). Myeloma cell BCMA expression pre- and post-GSK2857916 is not available. He then started pembrolizumab, lenalidomide, and dexamethasone in late October 2016, achieving a durable MR that was ongoing when he opted to enroll on a different BCMA CAR T-cell (P-BCMA-101, CD3ζ/4-1BB domains, transposon-based vector) phase 1 trial.9 In April 2018, he received 1.64 × 108 CAR T cells following cyclophosphamide and fludarabine conditioning and achieved a partial response that is ongoing 12 months post-CAR T-cell infusion, without any subsequent therapy. Bone marrow myeloma cell percentage decreased from 65% preinfusion to 15% on day 79. Assessment of BCMA expression by flow cytometry demonstrated BCMA expression on most of the myeloma cells pre-CAR T-cell infusion, with ongoing BCMA expression on residual cells posttreatment (Figure 1E).
Methods
The clinical trials referenced in this report were registered at www.clinicaltrials.gov as #NCT02064387, #NCT02546167, and #NCT03288493 and were approved by the respective institutional review boards and scientific committees of the authors’ institutions (University of Pennsylvania and Memorial Sloan-Kettering Cancer Center). Informed consent was obtained from both patients before trial enrollment per the Declaration of Helsinki. Flow cytometry and immunohistochemistry assessment for BCMA expression was performed as described6,8 and as per Figure 1.
Results and discussion
To our knowledge, these are the first reported cases of patients who progressed on a prior BCMA-targeted therapy (both of which were given at clinically active doses)3,6 and subsequently tolerated and responded to a different BCMA-targeted modality, including patient 1, who received these therapies sequentially without intervening therapy. These were patients with limited remaining treatment options who benefited because they had trial options that did not exclude prior BCMA-directed therapy. This is especially important in the heavily relapsed/refractory setting, in which BCMA CAR T cells, despite their high response rates, do not lead to long-term durable remissions in the majority of cases.4,6,10 The long-term response durability of anti-BCMA ADCs and bispecific antibodies remains unknown at this time.
The differing mechanisms of action between ADCs and CAR T cells also support consideration of sequential use. CAR T-cell therapy by definition is dependent on patient T-cell function; in fact, premanufacturing T-cell phenotype and other parameters of “fitness” may be predictive of in vivo CAR T-cell expansion and clinical response.6,11 In contrast, ADCs work primarily through T cell–independent mechanisms. For GSK2857916, these mechanisms include direct multiple myeloma cell apoptosis via internalization and release of the MMAF toxin, antibody-dependent cellular cytotoxicity, via binding to Fc receptors on natural killer cells and monocytes, and inhibition of pro-survival and proliferation signals to multiple myeloma cells through blockade of the BCMA receptor.12,13 The optimal sequencing of these modalities remains unknown, stressing a need for validation of predictive biomarkers of response and resistance that may aid with selection of the best modality for a given patient.
An interesting feature of patient 2, who was refractory to proteasome inhibitors, immunomodulatory drugs, and monoclonal antibodies, is that after progressing on the ADC, he had relatively prolonged periods of disease control (roughly 18 months on pembrolizumab, lenalidomide, dexamethasone, and 12+ months so far after BCMA CAR T cells) despite only modest paraprotein responses. Although this may simply reflect a more indolent biology of his myeloma, it raises the question of whether there was some benefit to the particular sequence of therapies he received. For example, GSK2857916 has also been postulated to induce immunogenic cell death,12 potentially priming an endogenous anti-myeloma immune response which theoretically could have been augmented by subsequent PD-1 inhibitor therapy. In addition, given the long half-life of monoclonal antibodies, pembrolizumab was likely still present in circulation (and perhaps still bound to PD-1 on T cells) at the time of leukapheresis and CAR T-cell manufacturing, which may have had a favorable effect on the final CAR T-cell product.14 All of this is speculative, but supports exploring combinations of ADCs and CAR T cells with PD-1/PD-L1 inhibitors, as in ongoing studies (eg, NCT03848845, NCT02706405). A final point is that neither patient achieved a very deep response to either BCMA-directed therapy; whether their experience can be extrapolated to patients who achieve a CR and/or minimal residual disease-negative state and then later relapse remains to be seen.
In sum, these cases demonstrate that BCMA-directed ADCs and CAR T cells can have clinical activity in patients who progressed on a prior BCMA-directed therapy. Prospective testing of each of these modalities, as well as BCMA-directed bispecific antibodies, in patients previously exposed to BCMA-directed therapy appears warranted. Careful correlative analysis of the dynamics of BCMA expression pre- and posttreatment may ultimately help determine optimal patient populations and sequence of administration for these agents, and thus additional comparative testing and standardization of BCMA detection methods (eg, flow cytometry vs immunohistochemistry)2 should also be a priority for the field.
Acknowledgments
This work was supported by the National Institutes of Health, National Cancer Institute (grant 1P01CA214278) (A.D.C., A.L.G., and S.F.L.); the Conquer Cancer Foundation of ASCO Career Development Award (A.L.G.); National Institutes of Health, National Cancer Institute (grants P30 CA008748 and P50 CA192937) (A.D., N.L., and A.M.L.); and the Sawiris Family Foundation and Parker Institute for Cancer Immunotherapy (A.M.L.).
Authorship
Contribution: A.D.C. and A.M.L. designed the research, performed the research, analyzed data, and wrote the manuscript; A.L.G., S.F.L., A.D., C.M., and M.S. performed the research and analyzed data; D.T.V. and N.L. performed the research; and all authors provided feedback and approved the manuscript.
Conflict-of-interest disclosure: A.D.C., A.L.G., and S.F.L. have received research support from Novartis and have had intellectual property licensed by the University of Pennsylvania to Novartis. A.D.C. has consulted for GlaxoSmithKline. A.D. has received personal fees from Roche, Corvus Pharmaceuticals, Physicians' Education Resource, Seattle Genetics, Peerview Institute, Oncology Specialty Group, Pharmacyclics, Celgene, and Novartis and research grants from Roche. C.M. and M.S. are employees of Poseida. The remaining authors declare no competing financial interests.
The current affiliation for N.L. is Janssen, Raritan, NJ.
Correspondence: Adam D. Cohen, Abramson Cancer Center, University of Pennsylvania, 3400 Civic Center Blvd, PCAM 12-South, 12-175, Philadelphia, PA 19104; e-mail: adam.cohen@uphs.upenn.edu.

