

Key Points
Increasing NO content in SSRBCs decreases epinephrine-activated SSRBC binding to the endothelium and leukocytes, and prevents vaso-occlusion.
Banked AARBCs stored for 30 days can increase SSRBC adherence and vaso-occlusion in vivo, unless these normal RBCs are loaded with NO.

Abstract
Sickle red blood cells (SSRBCs) are adherent to the endothelium, activate leukocyte adhesion, and are deficient in bioactive nitric oxide (NO) adducts such as S-nitrosothiols (SNOs), with reduced ability to induce vasodilation in response to hypoxia. All these pathophysiologic characteristics promote vascular occlusion, the hallmark of sickle cell disease (SCD). Loading hypoxic SSRBCs in vitro with NO followed by reoxygenation significantly decreased epinephrine-activated SSRBC adhesion to the endothelium, the ability of activated SSRBCs to mediate leukocyte adhesion in vitro, and vessel obstruction in vivo. Because transfusion is frequently used in SCD, we also determined the effects of banked (SNO-depleted) red blood cells (RBCs) on vaso-occlusion in vivo. Fresh or 14-day-old normal RBCs (AARBCs) reduced epinephrine-activated SSRBC adhesion to the vascular endothelium and prevented vaso-occlusion. In contrast, AARBCs stored for 30 days failed to decrease activated SSRBC adhesivity or vaso-occlusion, unless these RBCs were loaded with NO. Furthermore, NO loading of SSRBCs increased S-nitrosohemoglobin and modulated epinephrine’s effect by upregulating phosphorylation of membrane proteins, including pyruvate kinase, E3 ubiquitin ligase, and the cytoskeletal protein 4.1. Thus, abnormal SSRBC NO/SNO content both contributes to the vaso-occlusive pathophysiology of SCD, potentially by affecting at least protein phosphorylation, and is potentially amenable to correction by (S)NO repletion or by RBC transfusion.
Introduction
Sickle cell disease (SCD) morbidity is believed to arise from acute/chronic vaso-occlusion, caused largely by sickle red blood cell (SSRBC) adhesion to the vascular endothelial cells (ECs), leading to progressive multiorgan damage, affecting the heart, lungs, and kidneys, among other organs. SCD may involve a functional deficiency of bioactive nitric oxide (NO) derivatives,1-3 despite the fact that NO synthase activity is increased.4,5 NO is critical in the maintenance of vasodilation6,7 and is also a potent anti-inflammatory agent.8 Red blood cells (RBCs) act as hypoxia sensors that release bioactive NO derivatives, leading to NO-dependent vasodilation and increased blood flow to meet tissue oxygen demand.9 This action occurs after transfer of NO from heme iron (iron nitrosyl [Fe(NO)]) to cysteine thiol (forming S-nitrosothiol [SNO]). S-nitrosylation of the membrane anion transporter AE1 facilitates bioactive (S)NO release from RBCs.10 Bioactive NO species such as SNOs, whether precursor nitrite, SNOs, or the NO radical itself, are released and diffuse to the vessel wall, causing vasodilation.11
Abnormalities of NO-dependent regulation of vascular tone in SCD have been proposed to arise from consumption of NO by plasma-free hemoglobin (Hb).12 Hemolysis in SCD also releases erythrocyte arginase into the plasma, possibly lowering levels of arginine required for NO synthesis.13 However, SSRBCs are depleted in SNO stores, and HbS is less able to transfer NO from heme iron to cysteine thiol and form SNO-Hb.2 There is also reduced transfer of the NO moiety from SNO-HbS to the RBC membrane. Thus, the SSRBC may contribute to abnormal delivery of bioactive (S)NO in SCD and fail to elicit hypoxic vasodilation. The contribution of NO/SNO deficiency in SSRBCs to cell adhesion and vaso-occlusion has not previously been investigated. We therefore sought to test the hypothesis that SSRBC NO processing and bioactive NO delivery abnormalities are involved in SSRBC adhesion and vaso-occlusion.
RBC transfusion is a mainstay of treatment of some of the most severe complications of SCD, including acute chest syndrome and stroke. However, transfusion of stored RBCs, which are deficient in (S)NO,14 may be injurious and even raise mortality risk.15,16 Therefore, in addition to determining the effect of NO/SNO stores in SSRBCs on vaso-occlusion, we investigated how banked RBC NO/SNO deficiency influences the circulatory behavior of SSRBCs. These data could have broad implications regarding the outcomes of transfusion in both SCD and other settings.
Materials and methods
Endothelial cells
Pooled human umbilical vein ECs (HUVECs; ATCC), generated from multiple donors, were grown as previously described.17
Collection and preparation of RBCs
Blood samples were obtained from human participants after approval by Duke University's Institutional Review Board, and written informed consent was obtained from each participant. Blood samples were obtained from adult patients with SCD, 52% of whom were male, and from adult healthy control (“AA”) donors who matched patients with SCD in average age, sex, race, and blood type. Patients with SCD had not undergone transfusion for at least 3 months, had not experienced acute vaso-occlusive crises for 3 weeks, and were not taking hydroxyurea. Blood samples were collected into citrate tubes; stored leukoreduced RBCs were obtained from segments of banked RBC units (Duke Transfusion Service). Stored leukoreduced RBCs were in: AS-1 (40% of units), AS-3 (56%), or citrate-phosphate-dextrose-adenine (4%). Packed RBCs were separated as previously described in detail.18
RBC loading with NO
Aliquots of SSRBCs or stored banked RBCs were washed 3 times with phosphate-buffered saline (PBS) with 0.1 mM diethylenetriaminepentaacetic acid. RBCs (50% hematocrit) were partially deoxygenated under argon or helium for 30 minutes. Aqueous NO solution prepared from PROLI NONOate (Sigma-Aldrich) or NO gas (NO loading), or deoxygenated PBS (sham loading), was added to deoxygenated RBCs at a NO:heme ratio of 1:250. RBCs were then reoxygenated by exposure to air under gentle agitation and washed twice with air-equilibrated PBS with Ca2+ and Mg2+ before use.
Stability of RBC Hb-NO
Washed NO-loaded RBCs were serially assayed by using mercury-coupled photolysis-chemiluminescence (MPC)14 to assess the stability of total Hb-bound NO, bioactive SNO-Hb, and Hb[Fe]NO, and to determine whether intra-RBC NO adducts were sufficiently stable for use in in vivo experiments requiring that Hb-bound NO survive 60 to 90 minutes. Briefly, RBC lysate samples were desalted by using Sephadex G-25 (in PBS, pH 7.4, eliminating most sample nitrite; MilliporeSigma), then assayed by using photolytic liberation of NO bound to purified Hb in line with chemiluminescent NO quantification. Samples were treated in the presence or absence of inorganic mercury, which cleaves SNO groups. The MPC method has previously been shown to distinguish SNO-Hb and Hb[Fe]NO, measures both species essentially simultaneously, and is highly sensitive (detection limit of 1-3 pmol) for both species.19 Importantly, the MPC method avoids artifacts that are inherent in Hb-NO quantitation based on chemical reduction, such as triiodide.20 Under the conditions we used (UV-visible lamp, pH 7.4), photolysis of FeNO and SNO is highly efficient, and photolysis of residual nitrite is minimal.
Treatment of RBCs
Peripheral blood mononuclear cell separation and activation of adhesion by SSRBCs
Separation of peripheral blood mononuclear cells (PBMCs) from healthy donor blood and activation of PBMC adhesion to HUVECs by treated SSRBCs were performed as previously described in detail.23
In vitro adhesion assays
Assays of adhesion to HUVECs were performed in graduated-height flow chambers as described previously in detail.17
To determine whether NO radical released from SSRBCs during in vitro adhesion assays was critical for the effects observed, free HbA at 100 μM tetramer (HbA:HbS ratio of 10:1) was added to NO-loaded epinephrine-treated SSRBC suspensions (3 mL) to absorb any free NO, where indicated.
Mice
Animal work was approved by the Institutional Animal Care and Use Committee at Duke University. All surgery was performed under anesthesia achieved by intraperitoneal injection of 100 mg/kg of ketamine (Abbott Laboratory) and 10 mg/kg of xylazine (Bayer), and all efforts were made to minimize suffering. Female athymic homozygous nude (ν-/ν-) mice (Foxn1ν, formerly Hfh11ν), 8 to 12 weeks of age, were bred and housed at Duke University.
Window chamber surgery, RBC infusion, and intravital microscopy
Dorsal skin-fold window chamber surgery was performed on anesthetized mice. General anesthesia was achieved by intraperitoneal injection of 100 mg/kg of ketamine (Abbott Laboratory) and 10 mg/kg of xylazine (Bayer). A double-sided, titanium-frame window chamber was surgically implanted into the dorsal skin fold under sterile conditions by using a laminar flow hood. Surgery involved carefully removing the epidermal and dermal layers of one side of a dorsal skin fold, exposing the blood vessels of the subcutaneous tissue adjacent to the striated muscles of the opposing skin fold, and then securing the 2 sides of the chamber to the skin by using stainless steel screws and sutures. A glass window was placed in the chamber to cover the exposed tissue and secured with a snap ring. Subsequently, animals were kept at 32°C to 34°C until in vivo studies were performed 3 days postsurgery, at which time the animals had recovered from surgery.
Human RBC infusion into anesthetized animals with dorsal skin-fold window chambers and intravital microscopy were performed as previously described.18 Briefly, we infused fluorescently labeled NO-loaded or sham-loaded human RBCs, vehicle treated or treated with epinephrine (300 μL, hematocrit 50% in saline). SSRBC adhesion and blood flow dynamics were observed in subdermal vessels and recorded for at least 30 minutes by using 5×, 10×, and 20× magnification.
SSRBC adhesion was quantified by measurement of fluorescence intensity (pixels) of adherent fluorescently labeled SSRBCs in still images (20×) using Adobe Photoshop CS5 software (Adobe Systems). A color, single-channel fluorescent image (using equal-size images for each and for comparison of treatment conditions) was converted to “monochrome” for full control of how primary colors were converted correctly to grayscale. The image was then converted to a true 8-bit grayscale image suitable for fluorescence intensity quantification. Fluorescence intensity was quantified after 2 consecutive channel selections, to completely subtract both gray nonfluorescent areas and moving, dimly fluorescent SSRBCs, which appear less bright than adherent cells on still images.
In some experiments, fluorescently labeled vehicle-treated or epinephrine-treated human SSRBCs were admixed with nonlabeled sham-loaded or NO-loaded fresh or banked AARBCs, at a ratio of 4:1 ex vivo, before infusion.
RBC membrane ghost preparation and phosphopeptide enrichment
Nonradiolabeled membrane proteins of treated AARBCs and SSRBCs were prepared and separated by using mass spectrometry. They were then subjected to label-free quantitative phosphoproteomic analysis after phosphopeptide enrichment as described in detail in supplemental Material and methods, and previously.24,25
Statistical analysis
Results using sham and NO-treated RBCs were compared in paired fashion by using a single donor sample for each pair. Data were compared by using parametric analyses (GraphPad Prism 4 Software), including repeated and nonrepeated measures of analysis of variance. One-way and 2-way analyses of variance were followed by Bonferroni corrections for multiple comparisons. P < .05 was considered significant.
Results
Stability of Hb-bound NO in SSRBCs
Serial assays of total Hb-bound NO and SNO-Hb, and its precursor Hb[Fe]NO, in SSRBCs were performed before and after exposure to NO and compared with those in AARBCs. Total Hb-bound NO levels and the levels of basal SNO-Hb were greater in AARBCs than in SSRBCs (Figure 1A-B).10 However, basal levels of Hb[Fe]NO were comparable in SSRBCs and AARBCs (Figure 1C).2 After 1 minute of NO exposure, total Hb-NO and SNO-Hb increased significantly in both SSRBCs and AARBCs and were relatively stable for up to 2 hours (n = 3; P < .05 for sham-loaded vs NO-loaded SS and AARBCs, P > .05 for all subsequent time points compared with 1 minute). Although HbS[Fe]NO increased with NO exposure, there was little or no increase in HbA[Fe]NO. The minimal change in HbA[Fe]NO with NO exposure is most likely due to inadvertent cell aeration during processing in vitro, promoting the formation of SNO (SNO-Hb) immediately after the transient formation of HbA[Fe]NO. Accidental exposure to air during handling of relatively dilute Hb samples is typical and notoriously hard to avoid.11 HbS may be less susceptible because S-nitrosylation of HbS is disfavored more broadly.2 NO-induced SNO formation and its subsequent export may depend on the intermediate formation of Hb[Fe]NO. These findings indicate that a pool of stable NO adduct (both SNO-Hb and its precursor, Hb[Fe]NO) was present throughout subsequent experiments involving SSRBCs loaded with NO.
![Figure 1. Exposure of SSRBCs to NO increases formation of stable Hb-bound NO. Total NO bound to Hb (A), SNO-Hb (B), and Hb[Fe]NO (C) was assayed by photolysis-chemiluminescence before (sham) and at varying times (in minutes) after RBC exposure to aqueous NO solution (1:250 NO:Hb ratio). Results are expressed as number of moles (S)NO per moles of Hb. Hb-bound NO in SSRBCS (SS) after exposure to NO is stable for at least 3 hours. The mean and standard error of the mean (SEM) of 3 independent experiments are shown for each set of conditions. *P < .05 for both normal RBCs (AA) and SSRBCs (SS) vs the respective sham. None of the subsequent changes from 1 to 180 minutes was statistically significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/17/10.1182_bloodadvances.2019031633/4/m_advances031633f1.png?Expires=1769093445&Signature=qlxTBn9dgqgZfTv~bQxPT4gYHLc52mfBDEYBHugKMYi3SeR-G7Qcm0O7yXH4ccZxKcUTernNYTxcjwMJEO7~oyn2WQlnju56fkkp6QNyIAz-nlS22ihxcrnlGYuaNRFtqmGiG811Ws52vORgQvRYUsknT7hdl4MPYbFTcTTf3vU29CYYBS7doRB2-HJxTrJ5OjK72AgfhVjDFm-BzgDoQz9x8HPJPXcIg3rNhlJSKaWinpN4lA3P8YkSCucov2n-15JQTXH0VgJRhXzaunbQHVslAbymmVvSUk6hjGMLga38NGcJLR6d-q1CGZpJXZNpqJI84kexyvfrt7iXPkQd8g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Exposure of SSRBCs to NO increases formation of stable Hb-bound NO. Total NO bound to Hb (A), SNO-Hb (B), and Hb[Fe]NO (C) was assayed by photolysis-chemiluminescence before (sham) and at varying times (in minutes) after RBC exposure to aqueous NO solution (1:250 NO:Hb ratio). Results are expressed as number of moles (S)NO per moles of Hb. Hb-bound NO in SSRBCS (SS) after exposure to NO is stable for at least 3 hours. The mean and standard error of the mean (SEM) of 3 independent experiments are shown for each set of conditions. *P < .05 for both normal RBCs (AA) and SSRBCs (SS) vs the respective sham. None of the subsequent changes from 1 to 180 minutes was statistically significant.
Exposure of SSRBCs to NO increases formation of stable Hb-bound NO. Total NO bound to Hb (A), SNO-Hb (B), and Hb[Fe]NO (C) was assayed by photolysis-chemiluminescence before (sham) and at varying times (in minutes) after RBC exposure to aqueous NO solution (1:250 NO:Hb ratio). Results are expressed as number of moles (S)NO per moles of Hb. Hb-bound NO in SSRBCS (SS) after exposure to NO is stable for at least 3 hours. The mean and standard error of the mean (SEM) of 3 independent experiments are shown for each set of conditions. *P < .05 for both normal RBCs (AA) and SSRBCs (SS) vs the respective sham. None of the subsequent changes from 1 to 180 minutes was statistically significant.
The RBC NO-loading procedure was accompanied by a modest degree of hemolysis (<5%) but was followed routinely by RBC washing to remove residual free NO in solution. Under similar conditions, we had previously found that methemoglobin (metHb) does rise appreciably but to no more than ∼3% to 4% after NO addition to partially deoxygenated banked human RBCs.26 This low metHb yield likely reflects both the low NO:Hb stoichiometry we used and the presence of reductive RBC enzymatic systems such as methemoglobin reductase.
NO loading of SSRBCs inhibits adhesion to ECs in vitro
To determine whether a deficiency in the total NO/SNO content in SSRBCs2 is associated with abnormal SSRBC interaction with the endothelium, we supplemented human SSRBCs with NO. Cells were then washed, immediately treated with epinephrine, washed again, and assessed for adhesion to HUVECs. As previously described, vehicle-treated SSRBCs adhered to some degree to HUVECs at a shear stress of 1 dyne/cm2 (Figure 2A), whereas epinephrine enhanced SSRBC adhesion 2.7- ± 0.2-fold over baseline (P < .002).17 In contrast, and based on our previous extensive research, epinephrine was never able to significantly upregulate AARBC binding to ECs.17,18,23,24 Exposure of cells to NO before epinephrine treatment decreased epinephrine-stimulated SSRBC adhesion by 89% ± 6.4% (P < .01), whereas sham-loading (deoxygenation and then reoxygenation without NO) of SSRBCs failed to inhibit epinephrine-activated SSRBC adhesion (P > .05). Increasing SSRBC NO failed to affect the already low baseline SSRBC adhesion (data not shown). Thus, increasing RBC NO content reduced the ability of epinephrine to activate SSRBC adhesion.
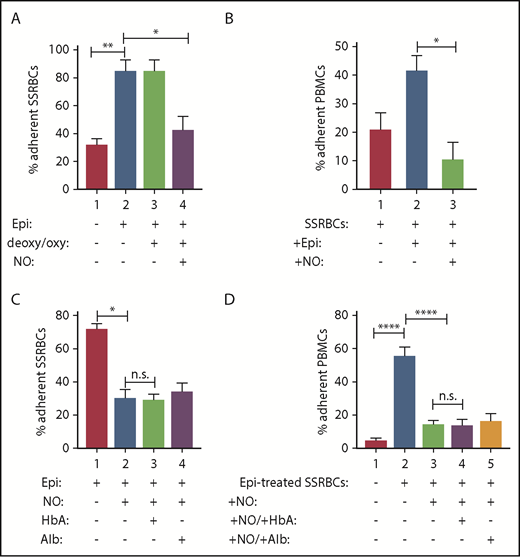
Exposure of SSRBCs to NO reduces both SSRBC adhesion and the ability of SSRBCs to activate mononuclear leukocyte adhesion. (A) NO loading of SSRBCs inhibits adhesion to ECs in vitro. SSRBCs were sham loaded (deoxygenation under helium/reoxygenation at room air) or NO loaded as described in "Materials and methods," followed by exposure to vehicle alone or to epinephrine (Epi) for 1 minute. Adhesion of SSRBCs to HUVECs was tested in intermittent flow condition assays. Results are presented as percent adherent SSRBCs at a shear stress of 1 dyne/cm2. Error bars show SEMs of 3 different experiments using blood samples from 3 different patient donors with SCD. **P < .002 for vehicle-treated vs Epi-treated; *P < .01 for sham-loaded Epi-treated vs NO-loaded Epi-treated. (B) NO prevents SSRBCs from inducing mononuclear leukocyte adhesion to ECs in vitro. SSRBCs were loaded with NO before epinephrine treatment. Adhesion of PBMCs pre-incubated with washed Epi-treated SSRBCs or washed NO-loaded Epi-treated SSRBCs were then assayed. Results are presented as percent adherent PBMCs at a shear stress of 1 dyne/cm2. Error bars show SEMs of 3 different experiments using blood samples from 3 different patient donors with SCD. *P = .0207 for PBMCs + Epi-treated SS vs PBMCs + NO-loaded Epi-treated SS. (C-D) The effect of SSRBC NO supplementation on adhesion in vitro is independent of release of NO itself. Adhesion of SSRBCs and PBMCs to HUVECs was tested in intermittent flow condition assays, and results are presented as percent adherent cells at a shear stress of 1 dyne/cm2. Error bars show SEMs of 3 different experiments using blood samples from 3 different patients with SCD for panels C and D, and 3 different healthy donors for panel D. (C) SSRBCs were treated with Epi for 1 minute, NO loaded followed by stimulation with Epi, NO loaded followed by stimulation with Epi and then incubation with free HbA, or NO loaded followed by stimulation with Epi then incubation with albumin (alb). *P = .0003 for Epi-treated vs NO-loaded Epi-treated. There was no statistically significant difference in adhesion of NO-loaded Epi-treated SSRBCs vs SSRBCs that had NO-loaded and Epi-treated and then incubated with either free HbA or alb. (D) SSRBCs were Epi-treated or NO loaded then Epi-treated, before coincubation with PBMCs in the presence of free HbA or alb. ****P < .0001 for PBMCs + Epi-treated SSRBCs vs PBMCs alone and PBMCs + NO-loaded Epi-treated SSRBCs vs PBMCs + Epi-treated SSRBCs. There was no statistically significant difference measured for adhesion of PBMCs + NO-loaded Epi-treated SSRBCs vs PBMCs + NO-loaded Epi-treated SSRBCs in the presence of free HbA or alb. n.s., not significant.
Exposure of SSRBCs to NO reduces both SSRBC adhesion and the ability of SSRBCs to activate mononuclear leukocyte adhesion. (A) NO loading of SSRBCs inhibits adhesion to ECs in vitro. SSRBCs were sham loaded (deoxygenation under helium/reoxygenation at room air) or NO loaded as described in "Materials and methods," followed by exposure to vehicle alone or to epinephrine (Epi) for 1 minute. Adhesion of SSRBCs to HUVECs was tested in intermittent flow condition assays. Results are presented as percent adherent SSRBCs at a shear stress of 1 dyne/cm2. Error bars show SEMs of 3 different experiments using blood samples from 3 different patient donors with SCD. **P < .002 for vehicle-treated vs Epi-treated; *P < .01 for sham-loaded Epi-treated vs NO-loaded Epi-treated. (B) NO prevents SSRBCs from inducing mononuclear leukocyte adhesion to ECs in vitro. SSRBCs were loaded with NO before epinephrine treatment. Adhesion of PBMCs pre-incubated with washed Epi-treated SSRBCs or washed NO-loaded Epi-treated SSRBCs were then assayed. Results are presented as percent adherent PBMCs at a shear stress of 1 dyne/cm2. Error bars show SEMs of 3 different experiments using blood samples from 3 different patient donors with SCD. *P = .0207 for PBMCs + Epi-treated SS vs PBMCs + NO-loaded Epi-treated SS. (C-D) The effect of SSRBC NO supplementation on adhesion in vitro is independent of release of NO itself. Adhesion of SSRBCs and PBMCs to HUVECs was tested in intermittent flow condition assays, and results are presented as percent adherent cells at a shear stress of 1 dyne/cm2. Error bars show SEMs of 3 different experiments using blood samples from 3 different patients with SCD for panels C and D, and 3 different healthy donors for panel D. (C) SSRBCs were treated with Epi for 1 minute, NO loaded followed by stimulation with Epi, NO loaded followed by stimulation with Epi and then incubation with free HbA, or NO loaded followed by stimulation with Epi then incubation with albumin (alb). *P = .0003 for Epi-treated vs NO-loaded Epi-treated. There was no statistically significant difference in adhesion of NO-loaded Epi-treated SSRBCs vs SSRBCs that had NO-loaded and Epi-treated and then incubated with either free HbA or alb. (D) SSRBCs were Epi-treated or NO loaded then Epi-treated, before coincubation with PBMCs in the presence of free HbA or alb. ****P < .0001 for PBMCs + Epi-treated SSRBCs vs PBMCs alone and PBMCs + NO-loaded Epi-treated SSRBCs vs PBMCs + Epi-treated SSRBCs. There was no statistically significant difference measured for adhesion of PBMCs + NO-loaded Epi-treated SSRBCs vs PBMCs + NO-loaded Epi-treated SSRBCs in the presence of free HbA or alb. n.s., not significant.
NO prevents SSRBCs from activating mononuclear leukocyte adhesion independently of NO release
Epinephrine-stimulated SSRBCs strongly activate PBMCs to subsequently adhere to HUVECs.23 We therefore determined the effect of NO loading of SSRBCs on SSRBC-stimulated PBMC adhesion. SSRBCs were treated with vehicle, epinephrine, or NO loading + epinephrine; they were then washed and coincubated with fluorescence-labeled normal PBMCs before assays of PBMC adhesion. Because only fluorescence-labeled PBMCs were visualized, quantitation of adherent PBMCs did not include any remaining unlabeled sickle leukocytes or SSRBCs. Incubation of PBMCs with vehicle-treated vs epinephrine-treated SSRBCs resulted in 21% ± 5.6% vs 42% ± 5.2% adherent PBMCs (shear stress 1 dyne/cm2) (Figure 2B). NO loading of SSRBCs blocked epinephrine-treated SSRBCs from inducing PBMC adhesion (P = .0207). Thus, although epinephrine-stimulated SSRBCs are potent activators of PBMC adhesion,23 increasing the SSRBC total NO/SNO levels by NO loading seems to inhibit the ability of SSRBCs to activate PBMC adhesion.
NO is also critical in the maintenance of vasodilation27,28 and modulates VLA-4 and Mac-1 function on the neutrophil cell surface, which is accompanied by changes in neutrophil adherence to the extracellular matrix.29 We therefore asked whether the effect of NO loading of SSRBCs on SSRBC adhesion (Figure 2A) and on activation of PBMC adhesion (Figure 2B) is dependent on an effect on endothelium or on an effect on the leukocytes. Free HbA (or albumin as a control) was added to NO-loaded SSRBCs and NO-loaded SSRBC-PBMC mixtures from a new cohort of patients to capture released SSRBC NO before and during adhesion assays, respectively. Again, epinephrine-treated SSRBCs were both strongly adherent (Figure 2C) and able to significantly increase PBMC adhesion (P < .0001) (Figure 2D). However, epinephrine-stimulated SSRBC adhesion and epinephrine-activated SSRBC stimulation of PBMC adhesion were both inhibited by preloading the SSRBCs with NO (P <0.001), even when free HbA was added to the NO-loaded SSRBC suspensions. Comparable results were obtained when albumin was added to NO-loaded cell suspensions.
To exclude the possibility that a portion of the free HbA added to our cell–cell mixture was unable to block NO group transfer from SSRBCs to ECs or PBMCs due to HbA conversion to metHb, supernatants from our cell suspensions were assayed for metHb. We were unable to detect metHb in the supernatants tested (supplemental Figure 1).30 Possible traces of metHb could be present but would not prevent the majority of reduced Hb from scavenging NO. Moreover, NO signal (by chemiluminescence) generated from PROLI NONOate was essentially completely eliminated in the presence of HbA (supplemental Figure 2). Together, these data suggest that SSRBC NO/SNO content primarily affects processes within the RBCs themselves, and any responsible extracellular mediator, which could be a free Hb-invulnerable NO-derivative, was not consistent with NO itself.
Effect of NO loading of SSRBCs on in vivo adhesion and vaso-occlusion
These experiments were designed specifically to study the effect of NO replenishment on human SSRBC adhesion and vaso-occlusion in vivo in the absence of the potential effect of NO loading on non-human cells, including murine RBCs, endothelium, leukocytes, or platelets. The goal also was to characterize the effect of NO supplementation of human SSRBCs on in vivo microcirculatory behavior. We have previously shown that treatment of human SSRBCs with epinephrine ex vivo markedly increased the adhesion of infused SSRBCs to the vascular endothelium, leading to vaso-occlusion.18 Intravital microscopy performed immediately after fluorescently labeled human SSRBC infusion showed that vehicle-treated (without epinephrine) SSRBCs adhered only occasionally to vessels visible through the dorsal skin-fold window chamber implants in nude mice (Figure 3A-B). However, epinephrine-stimulated SSRBCs adhered to a large number of microvessels, forming microaggregates that occluded vessels with evident blood stasis (Figure 3C-E), confirming our previous data. In sharp contrast, NO-loaded, epinephrine-activated SSRBCs displayed only minimal adhesion and vaso-occlusion, resulting in improved RBC circulatory behavior (Figure 3F-H; supplemental Movie 1). Sham-loading (exposure to hypoxia/reoxygenation only) had no inhibitory effect on epinephrine-activated SSRBC adhesion and vaso-occlusion (Figure 3I-K; supplemental Movie 2).
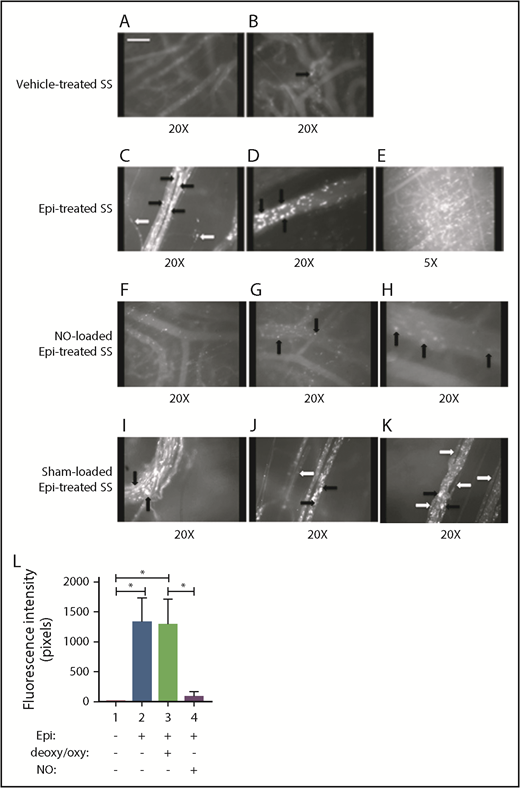
Replenishment of NO in SSRBCs reduces Epi-activated SSRBC adhesion to the endothelium and vessel occlusion in vivo. (A-K) Microscopic observations of postcapillary venules were conducted through implanted dorsal skin-fold window chambers after infusion of human SSRBCs into the tail vein of nude mice using 5× and 20× magnification. Vessels without adherent cells appear gray, due to the blurred fluorescence of rapidly moving SSRBCs. Infusion of vehicle-treated (n = 5; A-B), Epi-treated (n = 5; C-E), NO-loaded Epi-treated (n = 5; F-H), or sham-loaded Epi-treated (n = 5; I-K) human SSRBCs was performed. (A-E) Vehicle-treated human SSRBCs showed little adhesion to vessel walls (indicated by black arrows), whereas Epi-treated human SSRBCs showed marked adhesion to postcapillary venules, as indicated by black arrows, with intermittent vaso-occlusion, as indicated by white arrows. (F-K) In contrast, NO-loaded Epi-treated human SSRBCs displayed only minimal adhesion (indicated by black arrows) and no vaso-occlusion, whereas sham loading had no inhibitory effect on Epi-treated SSRBC adhesion (indicated by black arrows) and vaso-occlusion (indicated by white arrows). Scale bar, 50 μm. (L) Fluorescence intensity (pixels) represents fluorescence-labeled human SSRBC adhesion to vessel walls quantified by examining movies produced using 20× magnification. The values of segments of vessels analyzed were averaged among groups of animals to represent the mean fluorescence intensity. Error bars show SEM of 5 different experiments for each treatment. *P < .05 for either Epi-treated or sham-loaded Epi-treated vs vehicle-treated, and NO-loaded Epi-treated vs sham-loaded Epi-treated.
Replenishment of NO in SSRBCs reduces Epi-activated SSRBC adhesion to the endothelium and vessel occlusion in vivo. (A-K) Microscopic observations of postcapillary venules were conducted through implanted dorsal skin-fold window chambers after infusion of human SSRBCs into the tail vein of nude mice using 5× and 20× magnification. Vessels without adherent cells appear gray, due to the blurred fluorescence of rapidly moving SSRBCs. Infusion of vehicle-treated (n = 5; A-B), Epi-treated (n = 5; C-E), NO-loaded Epi-treated (n = 5; F-H), or sham-loaded Epi-treated (n = 5; I-K) human SSRBCs was performed. (A-E) Vehicle-treated human SSRBCs showed little adhesion to vessel walls (indicated by black arrows), whereas Epi-treated human SSRBCs showed marked adhesion to postcapillary venules, as indicated by black arrows, with intermittent vaso-occlusion, as indicated by white arrows. (F-K) In contrast, NO-loaded Epi-treated human SSRBCs displayed only minimal adhesion (indicated by black arrows) and no vaso-occlusion, whereas sham loading had no inhibitory effect on Epi-treated SSRBC adhesion (indicated by black arrows) and vaso-occlusion (indicated by white arrows). Scale bar, 50 μm. (L) Fluorescence intensity (pixels) represents fluorescence-labeled human SSRBC adhesion to vessel walls quantified by examining movies produced using 20× magnification. The values of segments of vessels analyzed were averaged among groups of animals to represent the mean fluorescence intensity. Error bars show SEM of 5 different experiments for each treatment. *P < .05 for either Epi-treated or sham-loaded Epi-treated vs vehicle-treated, and NO-loaded Epi-treated vs sham-loaded Epi-treated.
Quantitative analysis of fluorescence intensity, the indicator of adherent cells in mice infused with epinephrine-activated SSRBCs, showed increased fluorescence intensity by 269-fold (n = 5) (Figure 3C-E,L), compared with that in animals infused with vehicle-treated SSRBCs (n = 5) (P < .05) (Figure 3A-B,L). In contrast, loading SSRBCs with NO before exposure to epinephrine decreased fluorescence intensity by 86% ± 13.6% and 96% ± 3.1% (n = 5) (Figure 3F-H,L) compared with animals infused with epinephrine-activated SSRBCs (Figure 3C-E,L) or sham-loaded epinephrine-activated SSRBCs (Figure 3I-L), respectively (P < .05 for both differences). Together, these data suggest that endogenous SSRBC NO/SNO plays a critical role in RBC adhesion and vaso-occlusion in vivo.
Effect of banked AARBCs on SSRBC adhesion and vaso-occlusion in vivo
Although transfusion is often used in SCD, little is known about the effects of stored AARBCs on circulating SSRBCs. Stored AARBCs are deficient in (S)NO, and vasodilation by stored RBCs was significantly depressed.14 However, these (S)NO-deficient cells may gradually become repleted in NO/SNO while traversing the hypoxic microvascular environment after transfusion. We therefore hypothesized that the (S)NO deficiency of banked AARBCs, even if only transient, may be either deleterious or insufficiently beneficial in the setting of SCD. To determine the effects of banked AARBCs on SSRBC adhesion in vivo, fresh or banked AARBCs stored for 14 or 30 days were admixed with epinephrine-activated SSRBCs (4:1 ratio SSRBCs:AARBCs) ex vivo before infusion into the animals. Once again, vehicle-treated SSRBCs alone showed minimal adhesion to vessels and no visible vaso-occlusion (Figure 4A-B). However, although epinephrine-stimulated SSRBCs adhered avidly, promoting vaso-occlusion (Figure 4C-D; supplemental Movie 3), admixture with either fresh AARBCs separated from freshly drawn blood (immediately postacquisition) (Figure 4E-G) or AARBCs stored for 14 days (Figure 4H-J; supplemental Movie 4) reduced the adhesion of epinephrine-activated SSRBCs, inducing little to no vascular blockade. In contrast, admixture with AARBCs stored for 30 days failed to decrease adherence of epinephrine-activated SSRBCs or vaso-occlusion (Figure 4K-M; supplemental Movie 5).
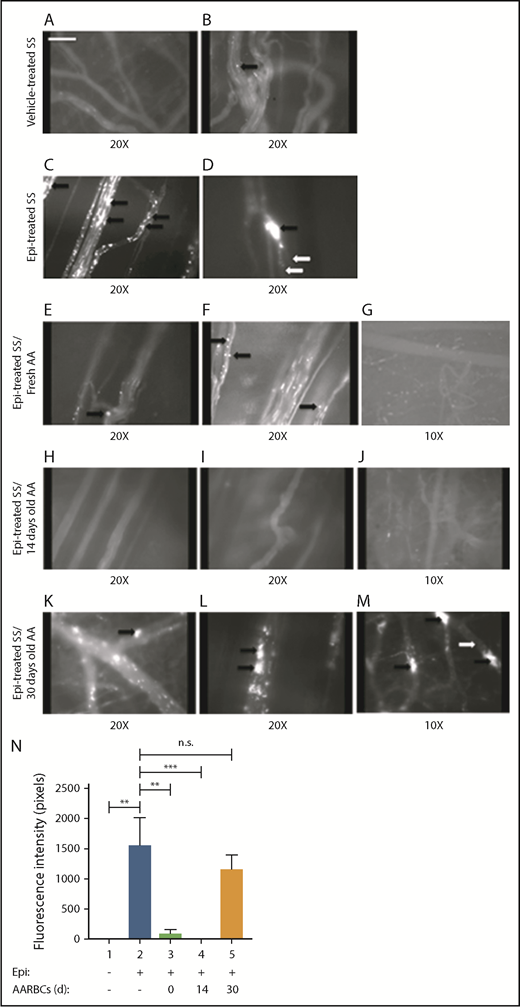
Banked normal AARBCs stored for 30 days failed to reduce SSRBC adhesion to the vascular endothelium and vaso-occlusion in vivo. (A-M) Microscopic observations of postcapillary venules were conducted by using 10× and 20× magnification through implanted dorsal skin-fold window chambers after infusion of human SSRBCs into the tail vein of nude mice. Vessels without adherent cells appear gray, due to the blurred fluorescence of rapidly moving SSRBCs. Mice received infusion of vehicle-treated SSRBCs, Epi-treated SSRBCs, or Epi-treated SSRBCs mixed with fresh AARBCs or AARBCs stored for 14 or 30 days. Vehicle-treated SSRBCs adhered minimally to the endothelium in vivo (A-B), whereas Epi dramatically increased SSRBC adhesion (indicated by black arrows) and vaso-occlusion (indicated by white arrows) (C-D). Infusion of either fresh (“0 day”) AARBCs mixed with epi-treated SSRBCs (E-G), or AARBCs stored for 14 days mixed with Epi-treated SSRBCs (H-J), dramatically reduced SSRBC adhesion and vascular stasis compared with Epi-stimulated SSRBCs alone. In contrast, admixture with AARBCs stored for 30 days (K-M) failed to decrease adherence of Epi-activated SSRBCs to the endothelium (indicated by black arrows) and vascular occlusion (indicated by white arrows). (N) Fluorescence intensity (pixels) representative of human SSRBC adhesion to vessels. Movies (20× magnification) were used to quantify fluorescence intensity induced by adherent human SSRBCs in animals infused with fluorescence-labeled SSRBCs treated as described in panels A-M (n = 5 for each treatment). The values were averaged among groups of animals to represent the mean fluorescence intensity. Error bars show SEM. **P < .05 for Epi-treated SS vs vehicle-treated SS, and Epi-treated SS + fresh AA vs Epi-treated SS. ***P < .05 for Epi-treated SS + 14-day-old AARBCs vs Epi-treated SS.
Banked normal AARBCs stored for 30 days failed to reduce SSRBC adhesion to the vascular endothelium and vaso-occlusion in vivo. (A-M) Microscopic observations of postcapillary venules were conducted by using 10× and 20× magnification through implanted dorsal skin-fold window chambers after infusion of human SSRBCs into the tail vein of nude mice. Vessels without adherent cells appear gray, due to the blurred fluorescence of rapidly moving SSRBCs. Mice received infusion of vehicle-treated SSRBCs, Epi-treated SSRBCs, or Epi-treated SSRBCs mixed with fresh AARBCs or AARBCs stored for 14 or 30 days. Vehicle-treated SSRBCs adhered minimally to the endothelium in vivo (A-B), whereas Epi dramatically increased SSRBC adhesion (indicated by black arrows) and vaso-occlusion (indicated by white arrows) (C-D). Infusion of either fresh (“0 day”) AARBCs mixed with epi-treated SSRBCs (E-G), or AARBCs stored for 14 days mixed with Epi-treated SSRBCs (H-J), dramatically reduced SSRBC adhesion and vascular stasis compared with Epi-stimulated SSRBCs alone. In contrast, admixture with AARBCs stored for 30 days (K-M) failed to decrease adherence of Epi-activated SSRBCs to the endothelium (indicated by black arrows) and vascular occlusion (indicated by white arrows). (N) Fluorescence intensity (pixels) representative of human SSRBC adhesion to vessels. Movies (20× magnification) were used to quantify fluorescence intensity induced by adherent human SSRBCs in animals infused with fluorescence-labeled SSRBCs treated as described in panels A-M (n = 5 for each treatment). The values were averaged among groups of animals to represent the mean fluorescence intensity. Error bars show SEM. **P < .05 for Epi-treated SS vs vehicle-treated SS, and Epi-treated SS + fresh AA vs Epi-treated SS. ***P < .05 for Epi-treated SS + 14-day-old AARBCs vs Epi-treated SS.
Admixture of epinephrine-stimulated SSRBCs with fresh (n = 5) or banked (14 days old, n = 5) AARBCs decreased fluorescence intensity by 94% and 99.5%, respectively, compared with epinephrine-stimulated SSRBCs alone (n = 5, P < .05 for either fresh or 14-day-old AARBCs + epinephrine-treated SSRBCs vs epinephrine-treated SSRBCs) (Figure 4N). In contrast, 30-day-old stored AARBCs (n = 5) failed to lower fluorescence intensity in the microvasculature (P > .05 for 30-day-old AARBCs + epinephrine-treated SSRBCs vs epinephrine-treated SSRBCs alone), suggesting that infusion of AARBCs stored ≥30 days may be unsuccessful in reducing SSRBC adhesion and vaso-occlusion.
Banked (30-day-old) AARBCs loaded with NO decreased SSRBC adhesion and vaso-occlusion in vivo
The deleterious effects of transfusion of banked AARBCs have been previously postulated to be related at least in part to SNO deficiency in stored AARBCs, a lesion also postulated to be involved in vaso-occlusive pain crises.2,14 We therefore attempted to attenuate vaso-occlusion induced by epinephrine-stimulated SSRBCs by loading 30-day stored AARBCs with NO before admixture with activated SSRBCs. Epinephrine-treated SSRBCs adhered strongly to the endothelium, promoting vaso-occlusion (Figure 5A-B). Although coinfusion of 30-days-stored AARBCs failed to reduce SSRBC adhesion or vaso-occlusion (Figures 5A,C-E; supplemental Movie 6), admixture with NO-loaded 30-days-stored AARBCs abrogated epinephrine-stimulated SSRBC adhesion and vaso-occlusion (Figure 5F-H; supplemental Movie 7), improving the circulation of epinephrine-treated SSRBCs. Fluorescence intensity was reduced by 95% when epinephrine-activated SSRBCs were mixed with NO-loaded stored AARBCs (n = 5) compared with epinephrine-treated SSRBCs (P < .005) (Figure 5I). In contrast, 30-days-stored AARBCs significantly increased (1.6-fold) the adhesion of epinephrine-stimulated SSRBCs. Our data suggest that transfusion of banked AARBCs supplemented with NO can attenuate SSRBC adhesion and vaso-occlusion.
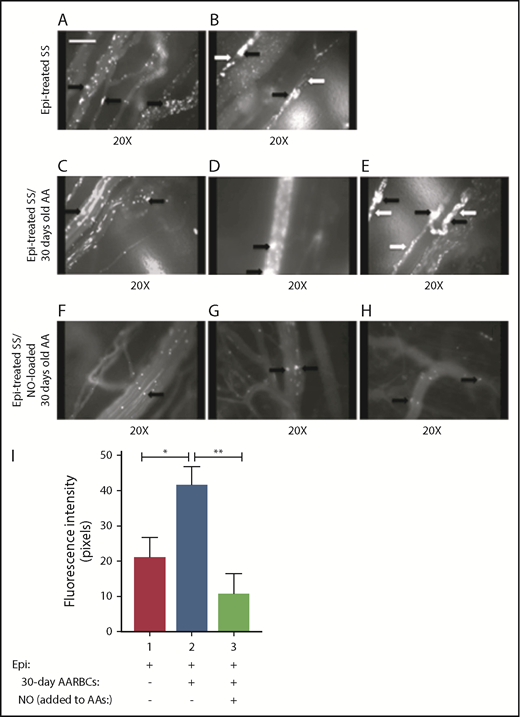
Loading banked (30 days old) AARBCs with NO decreased SSRBC adhesion and vaso-occlusion in vivo. (A-H) Inhibition of SSRBC adhesion with NO-loaded 30-day-old AARBCs was performed as described in "Materials and methods." Epi-treated SSRBCs (n = 5) exhibited marked adhesion to postcapillary venules, as indicated by black arrows, with intermittent vaso-occlusion as indicated by white arrows. Coinfusion with banked (30-day-old) AARBCs (n = 5) had no effect on Epi-treated SSRBC adhesion to postcapillary venules (indicated by black arrows) and vaso-occlusion (indicated by white arrows), whereas coinfusion with NO-loaded banked (30-day-old) AARBCs (n = 5) markedly inhibited adhesion of Epi-treated SSRBCs to postcapillary vessels (black arrows) with no vaso-occlusion. (I) Venule segments were analyzed by fluorescence intensity to quantify SSRBC adhesion. The values were averaged among groups of animals (n = 5 for each treatment) to represent the mean fluorescence intensity. Error bars show SEM of 5 different experiments. *P = .0207 for Epi-treated SS + old AA compared with Epi-treated SS; **P = .0046 for Epi-treated SS + old AA vs Epi-treated SS + NO-loaded old AA.
Loading banked (30 days old) AARBCs with NO decreased SSRBC adhesion and vaso-occlusion in vivo. (A-H) Inhibition of SSRBC adhesion with NO-loaded 30-day-old AARBCs was performed as described in "Materials and methods." Epi-treated SSRBCs (n = 5) exhibited marked adhesion to postcapillary venules, as indicated by black arrows, with intermittent vaso-occlusion as indicated by white arrows. Coinfusion with banked (30-day-old) AARBCs (n = 5) had no effect on Epi-treated SSRBC adhesion to postcapillary venules (indicated by black arrows) and vaso-occlusion (indicated by white arrows), whereas coinfusion with NO-loaded banked (30-day-old) AARBCs (n = 5) markedly inhibited adhesion of Epi-treated SSRBCs to postcapillary vessels (black arrows) with no vaso-occlusion. (I) Venule segments were analyzed by fluorescence intensity to quantify SSRBC adhesion. The values were averaged among groups of animals (n = 5 for each treatment) to represent the mean fluorescence intensity. Error bars show SEM of 5 different experiments. *P = .0207 for Epi-treated SS + old AA compared with Epi-treated SS; **P = .0046 for Epi-treated SS + old AA vs Epi-treated SS + NO-loaded old AA.
NO loading of epinephrine-stimulated SSRBCs decreased phosphorylation of multiple cell membrane proteins
A label-free quantitative phosphoproteomics analysis was applied to RBC membrane proteins prepared from RBCs. Each cell type was analyzed under each of the following conditions: untreated; sham loaded (deoxygenated and then reoxygenated as is necessarily done for NO loading) then vehicle treated; sham loaded then epinephrine treated; and NO loaded then epinephrine treated. The goal was to identify the effect of NO loading on phosphorylation of RBC membrane protein targets. For stringency, we required a ratio ≥1.7 (1.7-fold or greater increase) for phosphorylation of a given protein to be considered substantially increased by NO loading of RBCs. Similarly, a ratio ≤0.59 was considered adequate to conclude that phosphorylation of a protein significantly had decreased due to NO loading of RBCs. Proteins with a ratio of change in phosphorylation between 0.59 and 1.7 were eliminated from further consideration. Phosphorylation of Ser709 on the cytoskeletal protein 4.1 was 6-fold more abundant in SSRBCs vs AARBCs (P = .01) (Table 1). However, NO repletion of SSRBCs before epinephrine treatment increased by 2.64-fold, 1.7-fold, and 1.85-fold Ser709 phosphorylation within protein 4.1 (P = .019), Ser19 of pyruvate kinase isozymes R/L (P = .012), and Ser181 of E3 ubiquitin ligase UBR4 (P = .018), respectively. In contrast, sham loading or NO loading of AARBCs before epinephrine treatment, or sham loading of AARBCs or SSRBCs before vehicle treatment, had no effect on phosphorylation of these 3 proteins (supplemental Table 1).
Motif-specific phosphorylation affected by NO loading of SSRBCs
| Baseline (AA vs SSRBCs) | Effect of NO loading in SSRBCs | ||
|---|---|---|---|
| Protein description | Modified peptide sequence | Untreated SSRBCs vs AARBCs (P) | NO-loaded epinephrine-treated SSRBCs vs sham-loaded epinephrine-treated SSRBCs (P) |
| Protein 4.1 | RLS*THSPFR | 6-fold increase (.01) | 2.64-fold increase (.019) |
| Pyruvate kinase isozymes R/L | SWVSKS*QR | 0 | 1.70-fold increase (.012) |
| E3 ubiquitin-protein ligase UBR4 | TLSDVEDQKELASPVS*PELR | 0 | 1.85-fold increase (.018) |
| Baseline (AA vs SSRBCs) | Effect of NO loading in SSRBCs | ||
|---|---|---|---|
| Protein description | Modified peptide sequence | Untreated SSRBCs vs AARBCs (P) | NO-loaded epinephrine-treated SSRBCs vs sham-loaded epinephrine-treated SSRBCs (P) |
| Protein 4.1 | RLS*THSPFR | 6-fold increase (.01) | 2.64-fold increase (.019) |
| Pyruvate kinase isozymes R/L | SWVSKS*QR | 0 | 1.70-fold increase (.012) |
| E3 ubiquitin-protein ligase UBR4 | TLSDVEDQKELASPVS*PELR | 0 | 1.85-fold increase (.018) |
Fold change in phosphorylation for peptides as a function of NO loading of SSRBCs before epinephrine treatment are presented. Phosphorylation of all membrane proteins presented in the table was upregulated by NO loading of SSRBCs before epinephrine treatment. Phosphorylation sites are indicated by “*”. Data were generated by using blood samples from 4 different patients with SCD or 4 healthy donors.
Discussion
Previous studies have identified a deficiency of SNO-Hb and intrinsic defects in NO processing by SSRBCs, impairing formation of SNO and SNO-mediated vasodilation and vascular homeostasis.2,31 Most studies of NO in SCD have therefore focused on the effects of NO deficiency on the vasculature. Our findings pinpoint additional pathophysiologic effects of impaired RBC NO processing, promoting SSRBC adhesion, vaso-occlusion, and activation of PBMC adhesion. Our data also identify potential RBC membrane proteins regulated by the NO pathway and indicate that shortage of NO/SNO in SSRBCs affects cytoskeleton proteins and proteins involved in cytoskeletal organization. These results further suggest that deficiency in RBC NO/SNO likely affects processes within the RBCs associated with adhesive function.17,18,23
As we have previously shown, epinephrine-stimulated but not vehicle-treated human SSRBCs adhere avidly in vitro to human ECs17 and in vivo to murine vascular endothelium.18 This requirement for activation of SSRBCs with epinephrine to elicit robust adhesion is consistent with observations in humans, in whom SSRBCs most often circulate without causing symptomatic vaso-occlusion. However, NO loading before epinephrine stimulation of human SSRBCs dramatically reduced SSRBC–endothelial interactions and vaso-occlusion (Figure 6). In addition, NO supplementation also reduced the ability of epinephrine-stimulated SSRBCs to activate mononuclear leukocyte adhesion. Interestingly, the β2-adrenoceptors that epinephrine binds to in order to stimulate RBC adhesion can also couple to endothelial-type NOS in HUVECs,32 leading to NO production. RBCs reportedly contain endothelial NO synthase,33 but if any NO production occurred in response to epinephrine alone, it evidently had little functional antiadhesive effect compared with that seen when authentic, exogenous NO was added.
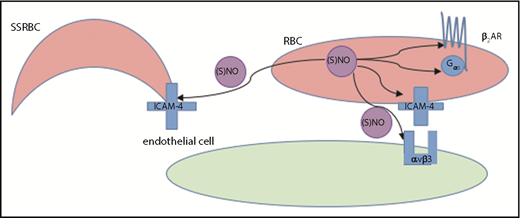
Model of NO-induced inhibition of SSRBC adhesion. RBC NO or SNO may modulate (eg, inhibit) intercellular adhesion via autocrine mechanisms such as by binding to reactive cysteine residues in the relevant G protein–coupled receptors such as the beta2-adrenergic receptor (β2AR), its downstream signaling partners such as the S-type G-protein (Gαs), or an adhesion receptor such as LW/ICAM-4. Alternatively, SNO (but not NO) exported from RBCs may inhibit adhesion by acting on adjacent cells in paracrine fashion; for example, during RBC-endothelial cell (or RBC-leukocyte) contact, possibly functionally modifying a counterreceptor such as α-v-β-3 (αvβ3) integrin, or when transfused AARBCs mix with native, activated SSRBCs.
Model of NO-induced inhibition of SSRBC adhesion. RBC NO or SNO may modulate (eg, inhibit) intercellular adhesion via autocrine mechanisms such as by binding to reactive cysteine residues in the relevant G protein–coupled receptors such as the beta2-adrenergic receptor (β2AR), its downstream signaling partners such as the S-type G-protein (Gαs), or an adhesion receptor such as LW/ICAM-4. Alternatively, SNO (but not NO) exported from RBCs may inhibit adhesion by acting on adjacent cells in paracrine fashion; for example, during RBC-endothelial cell (or RBC-leukocyte) contact, possibly functionally modifying a counterreceptor such as α-v-β-3 (αvβ3) integrin, or when transfused AARBCs mix with native, activated SSRBCs.
Reduced plasma bioactive NO has been attributed in part to NO consumption by free Hb released upon hemolysis.34,35 NO binds very rapidly to free deoxyhemoglobin in the plasma and forms a stable Hb(Fe2+)NO complex.36 NO also reacts with oxyhemoglobin, leading to the formation of metHb and NO3–. In addition, excessive superoxide generated by oxidized free Hb34,35 binds to NO to produce peroxynitrite, and may thereby not only contribute to a decrease in NO availability, but also to ongoing reactive oxygen species generation.37,38 Modestly increased levels of membrane and/or Hb-bound SNO achieved by supplementing SSRBCs with NO can reverse RBC vasoconstrictor activity ex vivo.2 In contrast to the original description of the β-chain Cys93-to-alanine mutant transgenic humanized mouse,39 recent work showed partial preservation of Hb-bound SNO in these mice, apparently in the form of fetal SNO-Hb.40,41 Nevertheless, HbF-SNO is not sufficient to prevent important cardiovascular phenotypes in mice, including susceptibility to myocardial infarction, and impaired peripheral blood flow and tissue oxygenation in hypoxia. Our data, including the lack of effect of the potent NO scavenger, soluble-free Hb, on the antiadhesive effect of NO loading, led us to hypothesize that the effect of (S)NO on SSRBCs depends at least partly on the actions of NO derivatives that affect the SSRBC adhesive phenotype and does not depend on the release of NO radical itself from RBCs. Rather, the effect, and the secondary impacts on endothelial cells and leukocytes, may depend on either autocrine action of NO or a derivative, directly or indirectly affecting proteins associated with RBC adhesion (Table 1), and/or the formation and/or export of a stable NO derivative, such as an SNO, which can resist scavenging by erythrocytic or free Hb and modify one of several protein targets.28
Possible protein and cellular targets of the antiadhesive effects of S-nitrosylation are illustrated in Figure 6. Indeed, in healthy human RBCs, blocking the export of S-nitroso-l-cysteine (L-CSNO) promoted adhesivity to ECs, an effect that could be overcome with L-CSNO in the medium, consistent with a basal antiadhesive role for RBC CSNO.42 Increasing levels of NO and its derivatives in SSRBCs regulate the effect of epinephrine on RBC adhesion and vaso-occlusion, at least in part by upregulating phosphorylation of the RBC cytoskeletal protein 4.1 at Ser709, as well as other proteins involved in cytoskeletal organization and regulation of integrin-mediated signaling, including E3 ubiquitin ligase UBR4 at Ser181, and pyruvate kinase isozymes R/L at Ser19.24 Studies have also shown that phosphatases may be inhibited by S-nitrosylation, as exemplified by the regulation of the protein tyrosine phosphatase 1b by SNOs.43 As such, NO loading of SSRBCs with subsequent increases in protein phosphorylation regulating the cytoskeleton arrangement may constitute a mechanism for controlling RBC adhesive function. Regulation of the effect of epinephrine on SSRBC adhesion by NO/SNO may also occur via other mechanisms yet to be identified. For instance, renitrosylation of human banked RBCs after 5 to 6 weeks of conventional storage attenuated the storage-induced increase in adhesivity and promoted spectrin S-nitrosylation.26 It is therefore possible that NO loading of SSRBCs regulates membrane protein phosphorylation as an allosteric consequence of the nitrosylation of protein cysteines,6,9 and both modifications may act synergistically to diminish SSRBC adhesion to ECs. The general question of whether RBC export of formed SNO is responsible for the antiadhesive effect of added NO could be addressed by using agents that inhibit CSNO export, as we have shown.42
Supplemental NO partially restores membrane SNO2 in SSRBCs, reflected in membrane-dependent properties such as RBC deformability and, possibly, in susceptibility to sickling. Not surprisingly, however, clinical studies have shown that neither the use of NO as inhaled NO gas44 nor supplementation of l-arginine, the NO precursor used by NO synthase, was clinically efficacious.9 In contrast, approaches that instead directly replenish NO/SNOs or their equivalent (nitrosylating species) in SSRBCs and resist inactivation by the cell-free Hb liberated in SCD are rational, because our data strongly argue that increased NO/SNO in SSRBCs reduces SSRBC adhesion and vaso-occlusion. In addition, alleviating oxidative stress in SCD may also replenish SSRBCs with bioactive NO. For instance, in SCD, the antioxidant N-acetylcysteine inhibits in vitro the formation of dense cells and irreversibly sickled cells, and restores glutathione levels to normal.45 In randomized clinical trials, administration of the antioxidant N-acetylcysteine to patients with SCD boosted whole blood glutathione levels, and decreased RBC phosphatidylserine exposure, vaso-occlusive episodes, and the frequency of pain crises.46,47
Our data further suggest that RBC transfusion, a mainstay of treatment of some severe complications of SCD, could be deleterious in some settings,15,16,48,49 as presumably the transfused banked cells require time to regain NO/SNO. Adhesion of activated SSRBCs and vaso-occlusion could be exacerbated when long-term, stored (≥30 days) normal RBCs are transfused, unless these cells are first (S)NO repleted just before transfusion. In contrast, fresh RBCs or RBCs stored for only 14 days were able to ameliorate vaso-occlusion when coinfused with SSRBCs. Because we did not study AARBCs stored for any period between 14 and 30 days, we do not know the precise timing of the functional, (S)NO-sensitive changes we documented. Previous reports showed that banked RBCs share with SSRBCs the characteristic of deficient SNO,14 suggesting that depletion in SNO during RBC storage may promote vasoconstriction by banked AARBCs, thus exacerbating SSRBC adhesion and vaso-occlusion in the transfused patient with SCD. Whereas we previously documented declines in RBC membrane total SNO content after only 3 hours of storage, the stability of individual protein–SNO adducts varies widely,50 and the SNO group on individual candidate proteins critical to RBC adhesive function may therefore remain bound for days or even weeks. It is also possible that storage-induced NO/SNO losses in AARBCs do not result in increased RBC adhesivity (or decreased ability to prevent SSRBC or PBMC adhesion) until other deficiencies accrue, such as the loss of antiadhesive intra-RBC (and exported) adenosine triphosphate, which occurs over weeks.51 This scenario would be consistent with our finding that NO loading of AARBCs was sufficient to enable coinfused AARBCs stored for 30 days to prevent SSRBC adhesion, mimicking both 0-days-stored (“fresh”) and 14-days-stored AARBCs. We did not measure NO adducts other than those bound to Hb, but found that the majority of the NO added bound to Hb. In addition, alternative reaction products such as nitrate or peroxynitrite would not be expected to leave the RBC once formed, except perhaps via intermediary conversion to SNO.9
Clinical evidence points to the potential importance of NO/SNOs in SCD. During vaso-occlusive crises, elevated NO metabolite levels are associated with lower clinical pain scores. This finding suggests that transfusion of banked RBCs stored for relatively shorter periods (eg, for ≤14 days), or enhancing intracellular NO/SNO levels in banked RBCs stored for longer periods (eg, >14 or 29 days), might decrease the severity of vaso-occlusive crises. The timing fluctuates among the various RBC “storage lesions,”14,26 and the clinical relevance of these biochemical and functional changes, is likely to vary additionally, as a function of the pathophysiology in the transfusate recipient. Together, our findings point to a new aspect of vaso-occlusion that results from alterations in NO/SNO processing and transfer within SSRBCs. Defects in NO/SNO in RBCs from patients with SCD as well as in banked RBCs suggest the therapeutic potential of modalities such as transfusion therapy that incorporates NO/SNO repletion of banked RBCs.
The full-text version of this article contains a data supplement.
Acknowledgments
This work was supported by a National Blood Foundation award to R.Z.; National Institutes of Health, National Heart, Lung, and Blood Institute, grants R01 HL137930 (R.Z.), R01 HL079915 (M.J.T.), and R01 HL107608 (M.J.T., T.J.M., and R.Z.) and National Institute of General Medical Sciences grant GM-113838 (T.J.M.); and VA Merit BX003478 (T.J.M.).
Authorship
Contribution: S.S performed window chamber surgery; D.A.R. performed experiments related to RBC S(NO) contents; R.Z. performed experiments related to in vitro and in vivo studies; M.B. performed in vitro adhesion experiments not included in the manuscript; H.Z. treated SSRBCs for the in vitro adhesion experiments not included in the manuscript; T.J.M., D.A.R., and R.Z. analyzed results; T.J.M. and R.Z. prepared figures; T.J.M., M.J.T., and R.Z. designed the research; T.J.M. and R.Z. wrote the paper; and all authors read and participated in revisions of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Rahima Zennadi, Division of Hematology and Duke Comprehensive Sickle Cell Center, Department of Medicine, Duke University Medical Center, 203 Research Dr, Durham, NC 27710; e-mail: zenna001@mc.duke.edu.

