

Key Points
An rFVIIa analog with increased procoagulant activity elicited anti-drug antibodies in some patients.
Redesigned rFVIIa analogs exhibit both desired functional activity and reduced immunogenicity risk.

Abstract
Vatreptacog alfa (VA), a recombinant activated human factor VII (rFVIIa) variant with 3 amino acid substitutions, was developed to provide increased procoagulant activity in hemophilia patients with inhibitors to factor VIII or factor IX. In phase 3 clinical trials, changes introduced during the bioengineering of VA resulted in the development of undesired anti-drug antibodies in some patients, leading to the termination of a potentially promising therapeutic protein product. Here, we use preclinical biomarkers associated with clinical immunogenicity to validate our deimmunization strategy applied to this bioengineered rFVIIa analog. The reengineered rFVIIa analog variants retained increased intrinsic thrombin generation activity but did not elicit T-cell responses in peripheral blood mononuclear cells isolated from 50 HLA typed subjects representing the human population. Our algorithm, rational immunogenicity determination, offers a broadly applicable deimmunizing strategy for bioengineered proteins.
Introduction
Hemophilia, a hereditary X-linked bleeding disorder, caused by a deficiency of factor (F) VIII (hemophilia A) or FIX (hemophilia B) is treated with replacement therapy using purified concentrates of FVIII or FIX. Development of neutralizing anti-drug antibodies (ADAs, also known as inhibitors) against FVIII or FIX is currently the most significant treatment complication encountered in treating hemophilia A and B. Activated prothrombin complex concentrate and activated recombinant FVII are used to bypass inhibitor activity and achieve hemostasis. Although the efficacy of each bypassing agent is high in controlled clinical trials, in acute situations, changes in either dose or bypassing product, or both, are commonly used to address life-threatening bleeding events. To expand bypassing treatment options by either achieving faster hemostasis after fewer doses or prolonging the interval between administrations, several modified recombinant activated human factor VIIa (rFVIIa) analogs with improved procoagulant activity or circulatory half-lives have been investigated.1-4 In 2012 and 2013, clinical trials for 2 different genetically engineered variants of blood coagulation FVIIa were terminated because some patients developed ADAs that may cross-react with endogenous FVIIa.5,6 Although the 2 FVIIa analogs were engineered with different mutations, both molecules were designed to produce better enzymatic activity over the existing therapy, a recombinant wild-type FVIIa (WT-rFVIIa) product. Unlike the bioengineered rFVIIa variants, there have been no reports of ADAs with the WT-rFVIIa, which has been used clinically for >2 decades.7
Previous studies have provided extensive evaluations of the immunogenicity of the bioengineered rFVIIa analog, vatreptacog alfa (VA), which resulted in ADAs in 11.1% of subjects enrolled in the phase 3 clinical trial.5 The VA variant differed from the WT-rFVIIa in 3 amino acid substitutions, V158D, E296V, and M298Q, achieving an up to 30- to 50-fold higher activity on activated platelets8 and good hemostasis in 93% of bleeds after just 1 dose.5 In a post hoc study,9,10 we demonstrated that the peptides with mutations at positions 296 and 298 are neo-epitopes, because they bind to human HLA-DRB1 alleles with high affinity, were identified on HLA-DRB1 molecules after processing by antigen presenting cells and resulted in T-cell activation.
The identification of high-risk HLA-DRB1 variants using immunogenicity prediction tools11-13 was supported by the HLA typing of patients in the clinical trial who developed immune responses. The putative neo-epitope generated by engineering VA was found to bind with high affinity to HLA variants identified in all patients who developed ADAs but in <50% of patients who did not develop ADAs, suggesting that peptide-HLA-DRB1 affinity is a good surrogate marker for clinical immunogenicity of VA.
In the study reported here, we used peptide-HLA-DRB1 affinity as a marker to develop an algorithm (rational immunogenicity determination [RID]) to deimmunize the bioengineered rFVIIa variant, VA. Several approaches to deimmunize protein therapeutics have been published.14-22 The computational and experimental approaches offer powerful strategies to deimmunize protein therapeutics. However, the redesigned deimmunized variants are difficult to evaluate because of the lack of suitable surrogate markers associated with clinical immunogenicity. Therefore, the evaluation of VA immunogenicity using in silico and ex vivo tools that we previously showed were associated with clinical immunogenicity is a first of its kind proof-of-principle experimental validation for a deimmunization strategy (Figure 1). The proposed de-immunized variants of VA exhibited decreased ex vivo CD4+ T-cell responses (to the baseline level) while simultaneously retaining the biological activity of the engineered variant.
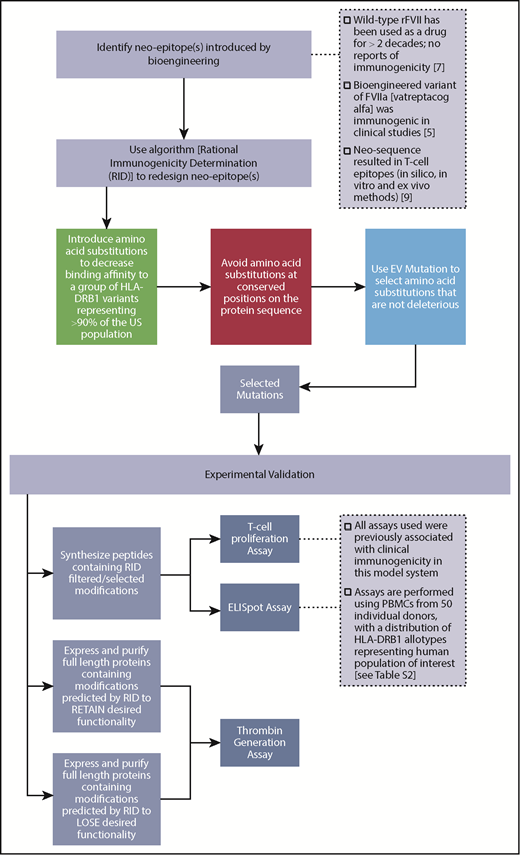
Overview of the computational and experimental strategies used in this study to design and evaluate an rFVIIa variant that has improved functional activity and lower immunogenicity risk.
Overview of the computational and experimental strategies used in this study to design and evaluate an rFVIIa variant that has improved functional activity and lower immunogenicity risk.
Materials and methods
Computational methods
We developed a computer algorithm, RID, to reduce immunogenicity risk while simultaneously preserving the desired functionality of an engineered protein therapeutic (supplemental Figure 1; supplemental Methods).
Protein expression
Wild-type FVII encompassing residues 61-466 (accession number P08709) was used as a starting template for site-directed mutagenesis for cloning the FVII variants. The expression constructs consisted of a cytomegalovirus promoter, a prolactin signal sequence, and an N-terminal protein A tag separated by a PreScission Protease cleavage site. The prolactin-protein A-FVII sequence was polymerase chair reaction amplified using primers containing PmeI restriction sites and cloned into the pJG lentiviral vector using InFusion-HD (Clontech, Mountain View, CA) per the manufacturer’s instructions. HEK 293T transfection, selection, and expression were carried out as previously described.23
Protein purification and activation
Protein variant purification was carried out as previously described.23 Fractions containing the monomeric form of FVII were combined and supplemented with 15 mM CaCl2, concentrated to 0.5 to 1 mg/mL, and left at ambient temperature for 48 to 96 hours (construct dependent) to allow for autoactivation to convert FVII to FVIIa.
Thrombin generation assay
The procoagulant activity of FVIIa variants was evaluated by a previously described thrombin generation assay24 in tissue factor (TF)– and procoagulant lipid (PL)–activated human plasma. FVIIa variant samples were serially diluted in Tris‐bovine serum albumin (BSA) buffer (pH 7.4) and added to a commercially available plasma from congenital hemophilia A patients (50% v/v final concentration [f.c.]; HRF Inc, Raleigh, NC) premixed with a platelet substitute in the form of phosphatidyl choline, phosphatidyl serine, and sphingomyelin phospholipid vesicles (4 μM f.c.; Phospholipid-TGT, Rossix, Mölndal, Sweden), and recombinant lipidated TF (0.5 pM f.c.; RecombiPlasTin, Instrumentation Laboratory Company, Lexington, MA). The experiments were initiated by rapid addition of plasma‐FVIIa variant mixtures (FVIIa concentrations: 0.03-1600 nM f.c.) to a mixture of calcium chloride (12.5 mM f.c.) and fluorogenic substrate for thrombin ZGGR‐AMC (800 μM f.c.; Bachem, Torrance, CA) in half-area transparent plates performed by a robotic 96‐channel pipettor (ViaFlo 96; Integra Biosciences, Bedford, MA). Fluorescence (product of thrombin substrate consumption) was recorded in a kinetic mode at 37°C using a Tecan Infinite F500 microplate reader (Tecan Group Ltd., Mannedorf, Switzerland) every 45 seconds for 1.7 hours. In‐house software was used to calibrate thrombin generation assay (using an internal thrombin calibrator CAT from Stago, Parsippany, NJ) and calculate thrombin peak height parameter. All experiments were performed using at least 2 independent protein preparations per FVIIa variant.
Clotting assay
Clotting assay was performed as the thrombin generation assay described here; however, clot formation was monitored through optical density (clot turbidity) at 412 nm in the absence of TF. Clot time was calculated automatically as the point when optical density reached one-half of its maximal value. Relative activities of FVIIa and FVIIa variants were calculated by comparing the clot times for several serial dilutions of each FVIIa variant sample (starting at 1600 nM) in a linear portion of the dose-response curve against a calibration curve prepared using the VA sample (ie, by definition, VA calibrator activity is 100%).
Isolation of PBMCs
Peripheral blood mononuclear cells (PBMCs) were obtained from healthy community subject buffy coats (from blood drawn within 24 hours), under consent, from commercial vendors. PBMC were isolated from buffy coats using Lymphocyte separation medium (Corning, Amsterdam, The Netherlands) density centrifugation and CD8+ T-cell depleted using CD8+ RosetteSep (StemCell Technologies Inc., London, United Kingdom).
T-cell proliferation assay
A cohort of 50 subjects was selected to best represent the number and frequency of HLA-DR and HLA-DQ allotypes expressed in the world population. PBMC were counted, viability assessed by acridine orange and propidium iodide using a Luna-FL Automated Cell Counter (Logos Biosystems, Annandale, VA), and suspended in AIM-V culture medium (Invitrogen, Paisley, United Kingdom) at 4 to 6 × 106 PBMC/mL. Bulk cultures were established for each subject where cells were added to a 24-well plate (Corning Life Science) along with peptide to give a final concentration of 5 µM. For each subject a clinically relevant positive control (cells incubated with exenatide [Bydureon, AstraZeneca, United Kingdom]) and a negative control (cells incubated with culture medium alone) were also included. An additional positive control used in the assay was Keyhole limpet hemocyanin. Proliferation of CD4+ T cells within the culture was measured on days 5, 6, 7, and 8 poststimulation by gently resuspending the cells and removal of 3 × 100 μL samples, which were transferred to a round bottomed 96-well plate and pulsed with 0.75 μCi/well tritiated thymidine (Perkin Elmer, Buckingham, United Kingdom). After 18 hours, the cultures were harvested onto filter mats (Perkin Elmer) using a TomTec Mach III cell harvester and counts per minute (cpm) for each well determined by Meltilex (Perkin Elmer) scintillation counting on a 1450 Microbeta Wallac Trilux Liquid Scintillation Counter (Perkin Elmer) in parallax, low background counting mode. All assays were performed in triplicate. Stimulation index (SI) was calculated by dividing the average counts per minute from peptide cultures by the average counts per minute in medium control cultures.
IL-2 ELISpot assay
ELISpot plates (Millipore, Watford, United Kingdom) were prewetted and coated with 100 µL/well interleukin-2 (IL-2) capture antibody (R&D Systems, Abingdon, United Kingdom) in phosphate-buffered saline (PBS). Plates were then washed twice in PBS, incubated overnight in blocking buffer (1% BSA in PBS) and washed in AIM-V medium. The cell density for each subject was adjusted to 4 to 6 × 106 PBMC/mL in AIM-V culture medium and cells were added to each well followed by peptide and controls. A mitogen control, Phytohemagglutinin (Sigma), was also included on each plate. After an 8-day incubation period, ELISpot plates were developed by sequential washing in dH2O and PBS (×3) before the addition of 100 µL of filtered biotinylated detection antibody (R&D Systems) in PBS/1% BSA. Following incubation at 37°C for 1.5 hours, plates were further washed in PBS (×3) and 100 µL of filtered streptavidin-AP (R&D Systems) in PBS/1% BSA was added for 1 hour (incubation at room temperature). Streptavidin-AP was discarded, and plates were washed in PBS (×3). A total of 100 µL BCIP/NBT substrate (R&D Systems) was added to each well and incubated for 30 minutes at room temperature. Spot development was stopped by washing the wells and the backs of the wells 3 times with dH2O. Dried plates were scanned on an Immunoscan Analyzer (Cellular Technology Limited, Cleveland, OH) and spots per well were determined using Immunoscan Version 5 software. SI was calculated by dividing the average number of spots observed in triplicate peptide cultures by the average number of spots observed in the medium control cultures.
Results
In silico profiling for HLA-protein affinity and protein stability
To ensure that we can provide adequate evaluation of deimmunization algorithm, we used the same nonclinical assessment tools previously associated with clinical immunogenicity of VA (Figure 1 and Lamberth et al9 ). To design deimmunized mutants of the VA protein, we developed a computational method, RID, based on minimization of the affinity of the neo-sequence for HLA-DRB1 variants (supplemental Figure 1). We applied RID to reengineer the residues 289 to 305 of the FVIIa sequence (ie, 7 residues up- and downstream of the neo-epitope at positions 296 and 298 in VA9 ) (supplemental Figure 2). The design space allowed for 323 possible amino acid substitutions. RID uses a tiered approach to design deimmunized, functional variants of VA.
We first used a promiscuity score25 to identify all substitutions that would show reduced binding to HLA-DRB1 molecules. The promiscuity score estimates the binding of peptides of interest to a set of HLA-DRB1 variants (supplemental Table 1) that contain ∼95% of the alleles in the world population and is the fraction of HLA-DRB1 variants in the dataset that the peptide binds to with high affinity, weighted for the frequency with which each HLA-DRB1 occurs in that population. Figure 2A is a binary plot that shows all substitutions that result in a decrease in the promiscuity score. Next, all amino acid substitutions that decrease in promiscuity scores are evaluated for sequence conservation. The highly conserved positions are considered functionally important and were not targeted during deimmunization (Figure 2B). The remaining amino acid substitutions were evaluated by EVmutation,26 an unsupervised statistical method that identifies deleterious variants and filters them out. The final set of amino acid substitutions (Figure 2C) represents those that are likely to result in decreased binding affinity for a broad spectrum of HLA variants and simultaneously retain desired biological function. To rank the potentially deimmunized epitopes, we computed the log-fold change in promiscuity scores compared with VA (Figure 2D). The scoring showed that mutations V299R and N301L provided only a minimal improvement in the promiscuity score over VA (Figure 2D).
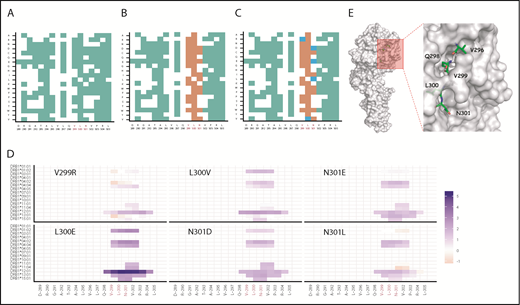
In silico design of deimmunized variants. Deimmunization strategy based on algorithm described in supplemental Figure 1. Deimmunization was applied to the neo-epitope identified in VA generated by introducing neo-sequences during protein engineering. (A) Positions in green indicate a predicted decrease in promiscuity scores for the given amino acid substitution. (B) Amino acid substitutions with a decreased promiscuity score were evaluated for conservation score; substitutions depicted in orange indicate substitutions with a conservation score ≥0.6. (C) The amino acid substitutions with a decrease in promiscuity score at nonconserved regions were evaluated using EVmutation; substitutions with a score ≥−0.25 are shown in blue. (D) The Log2 fold change of percent rank of predicted HLA-DRB1 binding compared with VA for each HLA allele. (E) Surface representation of FVII mutant (PDB ID: 3ELA). The potential mutation sites for deimmunization identified using RID are represented as sticks.
In silico design of deimmunized variants. Deimmunization strategy based on algorithm described in supplemental Figure 1. Deimmunization was applied to the neo-epitope identified in VA generated by introducing neo-sequences during protein engineering. (A) Positions in green indicate a predicted decrease in promiscuity scores for the given amino acid substitution. (B) Amino acid substitutions with a decreased promiscuity score were evaluated for conservation score; substitutions depicted in orange indicate substitutions with a conservation score ≥0.6. (C) The amino acid substitutions with a decrease in promiscuity score at nonconserved regions were evaluated using EVmutation; substitutions with a score ≥−0.25 are shown in blue. (D) The Log2 fold change of percent rank of predicted HLA-DRB1 binding compared with VA for each HLA allele. (E) Surface representation of FVII mutant (PDB ID: 3ELA). The potential mutation sites for deimmunization identified using RID are represented as sticks.
Finally, the mutations were mapped on the crystal structure of FVII (Figure 2E). This step was helpful because both conservation scores and EVmutation apply global rules and are not based on the specific structure of the protein being modified. For example, position 300 of WT-rFVIIa is buried in the protein core (Figure 2E), and the protein stability is likely to be affected by this substitution. Position 301, on the other hand, is solvent-exposed (Figure 2E) and substitutions at this position are unlikely to influence protein folding and functionality. Therefore, 2 functional deimmunizing mutations at position N301 (N301D and N301E) and 1 nonfunctional deimmunizing mutation at L300 (L300E) were used in the experimental validation.
T-cell activation by wild-type rFVIIa, vatreptacog alfa, and deimmunized peptides
Our previous evaluation of the neo-sequence (E296V/M298Q) introduced into VA demonstrated that the mutant peptides bind with high affinity to several HLA-DRB1 variants and elicit T-cell responses in an ex vivo assay using PBMC from healthy subjects.9 To evaluate the success of the deimmunization strategy we used 2 assays (a 3H-thymidine incorporation assay and an IL-2 ELISpot assay) that monitored expansion of CD4+ T cells in response to the peptides. In our computational strategy, we used a promiscuity score25 to address the diversity of the HLA repertoire; during experimental validation we used PBMCs from 50 HLA-typed subjects with a distribution of HLA variants comparable to that in the world population (supplemental Figure 1). PBMCs from 2 cohorts of 50 subjects each were exposed to WT-rFVII and VA sequences (experiment 1; supplemental Figure 4 and WT-rFVIIa, VA) and deimmunized sequences (experiment 2; Figure 3). Experiment 1 was used to ensure that the experimental strategy reproduced our previous finding9 that exposure VA peptides with the neo-epitope (E296V/M298Q) resulted in a significantly higher number of responders compared with exposure to WT peptides. Experiment 2 compared responses of cells from the same subjects to WT, VA and deimmunized rFVIIa sequences.
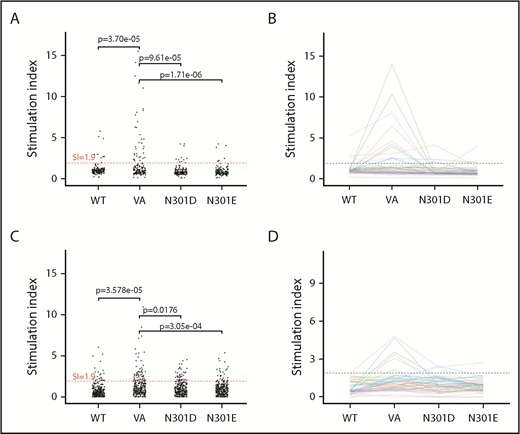
T-cell responses. T-cell proliferation assay. (A) Cells from 50 donors were subjected to a 3H-incorporation T-cell proliferation assay. PBMCs from each donor were stimulated with peptides derived from WT FVIIa, VA, and the mutants N301D and N301E. Day 8 SI values are shown. Comparisons between the groups were made using 1-sided Fisher's exact test on the hypotheses that there were (1) more responders to VA than WT and (2) fewer responders to either N301D or N301E than VA. The test compared the number of measurements that would be considered responders using a SI threshold of 1.9. Each dot represents a single measurement per donor. (B) The mean SI for each donor (3 replicates) was calculated. A mean SI > 1.9 (dotted line) is considered a positive responder. (C-D) IL-2 ELISpot assay. PBMCs from the same donors used in the T-cell proliferation assay were stimulated with the same peptides and IL-2 secreting cells were assessed by ELISpot assay. The analysis from panels A and B were repeated for the ELISpot assay.
T-cell responses. T-cell proliferation assay. (A) Cells from 50 donors were subjected to a 3H-incorporation T-cell proliferation assay. PBMCs from each donor were stimulated with peptides derived from WT FVIIa, VA, and the mutants N301D and N301E. Day 8 SI values are shown. Comparisons between the groups were made using 1-sided Fisher's exact test on the hypotheses that there were (1) more responders to VA than WT and (2) fewer responders to either N301D or N301E than VA. The test compared the number of measurements that would be considered responders using a SI threshold of 1.9. Each dot represents a single measurement per donor. (B) The mean SI for each donor (3 replicates) was calculated. A mean SI > 1.9 (dotted line) is considered a positive responder. (C-D) IL-2 ELISpot assay. PBMCs from the same donors used in the T-cell proliferation assay were stimulated with the same peptides and IL-2 secreting cells were assessed by ELISpot assay. The analysis from panels A and B were repeated for the ELISpot assay.
In the 3H-thymidine incorporation assay, PBMCs from a significantly higher number of subjects exhibited a SI > 1.9 when incubated with the neo-epitope derived from VA compared with incubation with the wild-type peptide (P = 3.7 × 10−5 (Figure 3A). The number of PBMC donors responding (SI > 1.9) to the deimmunized peptides (N301D and N301E) was significantly lower (P = 9.61 × 10−5 and P = 1.71 × 10−6, respectively) to that observed for the VA peptide (Figure 3A). Similarly, we found using the ELISpot assay, significantly higher numbers of responders for cells incubated with VA peptides compared with WT (P = 3.58 × 10−5) or deimmunized peptides (P = .0176 and P = 3.05 × 10−4) (Figure 3C). The average (n = 3) responses (SI) for cells from each of 50 subjects when incubated with WT, VA, and the deimmunized rFVIIa peptides in the 3H-thymidine incorporation (Figure 3B) and ELISpot (Figure 3D) assays are also shown. The responses of each subject in the 2 assays are shown in Figure 4 and used to determine a consensus response (ie, SI > 1.9 for both 3H-thymidine incorporation and IL-2 ELISpot assays). A consensus response was not found for any subject following treatment with the WT-rFVIIa peptide, and PBMCs from 2% and 0% of subjects were positive responders to the 2 deimmunized variants, N301D and N301E, respectively. In sharp contrast, PBMCs from 12% of subjects exhibited a consensus response to the peptide that incorporates the neo-epitope previously identified in VA. These data show that in assays used to monitor expansion of T cells in response to peptides, both deimmunized variants (N301D and N301E) are comparable to the WT-rFVIIa sequence (which is nonimmunogenic in the clinic) (ie, sequences designed using RID are likely to exhibit low immunogenicity risk).
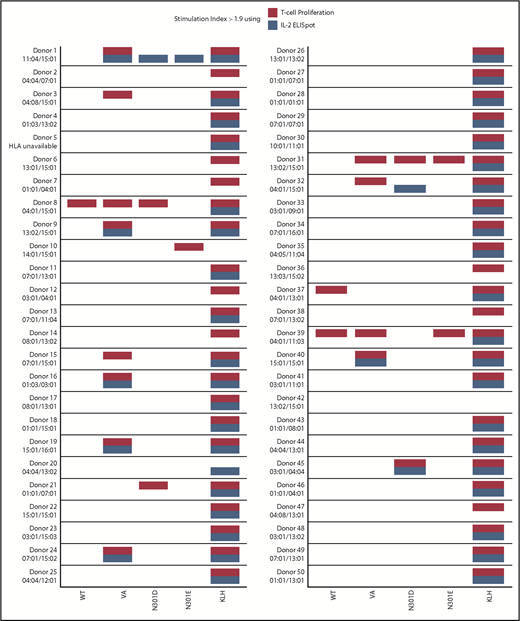
T-cell responses summary. A summary of donor responses, including high-resolution HLA-typing of the donors DRB1 allele, after restimulation with WT FVIIa, VA, and the mutants N301D and N301E peptides. Keyhole limpet hemocyanin (KLH) is included as a positive control. Only responders with SI > 1.9 (using 3 replicates) are shown.
T-cell responses summary. A summary of donor responses, including high-resolution HLA-typing of the donors DRB1 allele, after restimulation with WT FVIIa, VA, and the mutants N301D and N301E peptides. Keyhole limpet hemocyanin (KLH) is included as a positive control. Only responders with SI > 1.9 (using 3 replicates) are shown.
Altering protein sequence around neo-sequences to retain intrinsic activity
VA was bioengineered to improve its drug effect through a fivefold to 30-fold increase in the lipid surface-dependent enzymatic activity compared with WT-rFVIIa.8 To demonstrate that the deimmunization mutations did not disrupt the desired functionality, we expressed and purified WT-rFVIIa, VA, and the N301D and N301E variants on the VA background. The activities of the WT-rFVIIa, VA, and mutants were evaluated using the thrombin generation assay (TGA), which measures the coagulation (antibleeding) activity in the absence and presence of TF. Consistent with previous reports,8,27 VA showed increased intrinsic (TF-independent) activity compared with the WT-rFVIIa (Figure 5A-B). At 50 nM of FVIIa, significantly higher thrombin generation had been observed with VA, N301D, and N301E than with WT-rFVIIa in TF-independent experiments activated with PL concentrations from 1 to 16 µM (Figure 5C, left). In contrast, WT-rFVIIa and VA variants had comparable effects on TGA when activated with increasing concentrations of TF in the presence of 50 nM of FVIIa (Figure 5C, right).
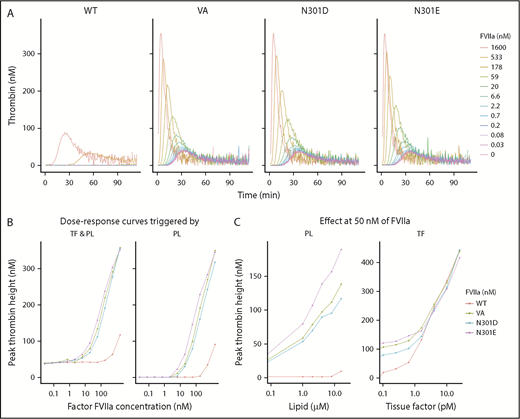
The functional activity of FVIIa variants. (A) Thrombin generation assays carried out using the following purified proteins: WT FVIIa, VA, and the mutants N301E and N301D using hemophilia A plasma (ie, plasma without functional FVIII). (B) The dose effect of FVIIa concentrations (from 0.03 to 1600 nM) for variants described in panel A. (A-B) The assay was carried out in the presence of phospholipid vesicles (4 µM) either in the presence (left) or absence (right) of TF (0.5 pM). (C) Effect of PL concentration (left, PL range: 0, 1-16 µM) and TF concentrations (right, TF range: 0, 0.1-25 pM) on thrombin generation by the FVIIa variants (FVIIa concentration: 50 nM) described in panel A.
The functional activity of FVIIa variants. (A) Thrombin generation assays carried out using the following purified proteins: WT FVIIa, VA, and the mutants N301E and N301D using hemophilia A plasma (ie, plasma without functional FVIII). (B) The dose effect of FVIIa concentrations (from 0.03 to 1600 nM) for variants described in panel A. (A-B) The assay was carried out in the presence of phospholipid vesicles (4 µM) either in the presence (left) or absence (right) of TF (0.5 pM). (C) Effect of PL concentration (left, PL range: 0, 1-16 µM) and TF concentrations (right, TF range: 0, 0.1-25 pM) on thrombin generation by the FVIIa variants (FVIIa concentration: 50 nM) described in panel A.
In all experiments that measured biological activity, VA, N301D, and N301E demonstrated comparable dose-effect curves. In an assay that measures time of blood plasma clotting supplemented with PL (supplemental Figure 5), using VA as a standard (VA = 100%), the activities of N301D, N301E, and WT-rFVIIa were 79% ± 5%, 150% ± 4%, and 4% ± 1%, respectively (mean ± standard deviation, n = 2 independent protein preparations) (Figure 6B).
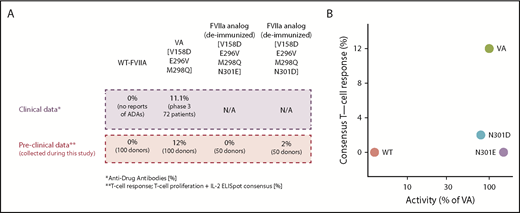
Molecules with desired functional activity and lower immunogenicity risk. (A) Correlation of preclinical data with clinical outcomes. (B) The relation between activity and T-cell response in all evaluated FVII variants. N301E and N301D exhibit desired characteristics for a therapeutic-protein (ie, low T-cell response and activity comparable to the VA). T-cell responses were measured using 2 independent experiments of 50 donors each. In experiment 1, PBMCs from 50 donors were exposed to WT-rFVII and VA sequences. In experiment 2, PBMCs from 50 donors were exposed to WT-rFVIIa, VA, and deimmunized sequences (N301E and N301D). Activities for each FVII variant are represented as a mean calculated from 2 independent protein preparations using VA as a standard (100%).
Molecules with desired functional activity and lower immunogenicity risk. (A) Correlation of preclinical data with clinical outcomes. (B) The relation between activity and T-cell response in all evaluated FVII variants. N301E and N301D exhibit desired characteristics for a therapeutic-protein (ie, low T-cell response and activity comparable to the VA). T-cell responses were measured using 2 independent experiments of 50 donors each. In experiment 1, PBMCs from 50 donors were exposed to WT-rFVII and VA sequences. In experiment 2, PBMCs from 50 donors were exposed to WT-rFVIIa, VA, and deimmunized sequences (N301E and N301D). Activities for each FVII variant are represented as a mean calculated from 2 independent protein preparations using VA as a standard (100%).
Finally, we evaluated RID for successfully weeding out deleterious mutations. Variants L300E, N301D, and N301E were predicted to be deimmunized. However, although mutants N301D and N301E were predicted to be functional, variant L300E was predicted to be functionally impaired (Figure 2C). Consistent with the prediction (Figure 2C), variants N301D and N301E showed tissue factor-independent activity comparable to VA (Figure 5A-B; supplemental Figure 6), whereas the variant L300E was nonfunctional (supplemental Figure 6).
Correlation between preclinical and clinical data
The results described here were obtained using methods previously validated for assessing the immunogenicity risk of rFVIIa variants and routinely used to determine the potency of FVIIa. In clinical experience and in clinical studies,7 there are no reports of ADAs with use of WT-rFVIIa. The results of the 2 independent ex vivo T-cell proliferation assays are consistent with the clinical experience. Moreover, 11.1% of subjects in a clinical study developed ADAs following infusion of VA, which is also consistent with results of the ex vivo T-cell proliferation assays because PBMC from 12% of subjects elicited a response (Figure 6A). Although the close match of the number of responsive subjects in the ex vivo assays and clinical study may be a coincidence, it is important that the ex vivo assays could identify a higher frequency of subjects responsive to VA compared with WT-rFVIIa. The ex vivo assays provided an additional level of confidence because they were conducted using a donor cohort sufficiently large (2 cohorts of 50 subjects each) to provide a broad and representative distribution of HLA-DRB1 variants (supplemental Figure 3; supplemental Table 2). Using this robust experimental system, which is associated with clinical immunogenicity, our evaluation of the 2 deimmunized sequences (N301E and N301D) shows that the deimmunized sequences were comparable to the WT-rFVIIa in terms of immunogenicity risk (Figure 6A). As shown in Figure 6B, the deimmunized analogs exhibit comparable activity to that that of VA while simultaneously having a lower risk of immunogenicity comparable to the WT-rFVIIa. Using RID, we reengineered FVIIa variants that retain increased intrinsic activity of VA without eliciting T-cell responses (Figure 6B).
Discussion
Humoral mechanisms mediate most of the adverse effects resulting from elicitation of an immune response to a therapeutic protein product and circulating ADAs are the chief criterion for defining an immune response to therapeutic proteins. Development of ADAs can affect safety or efficacy11,28-30 of a therapeutic protein. Particularly, ADAs that cause cross-reactive neutralization of endogenous proteins mediating critical functions pose a serious safety risk. Incidence of ADAs is often rare and requires prolonged exposure. In small subpopulations, protein immunogenicity may not be recognized until the commencement of large expensive phase 3 clinical studies5 or even after approval.31-35 This late-stage failure of drugs adds considerable economic risk for the biotechnology industry. More important, the potential consequences of immunogenicity for a subject in a clinical study range from loss in quality of life36 to a life-threatening situation.28 In recent years, immunogenicity-related issues have been exacerbated by the routine use of protein engineering. High-throughput technologies for generating and assaying protein variants allow for selecting variants that are likely to have properties that are optimal for a drug. The drug development pipeline suggests that most new therapeutic proteins do not use WT sequences but are genetically or chemically engineered to improve the serum half-life, stability, activity, binding properties, and so on.27,37,38 The changes introduced in the primary structure, which do not exist in nature, are potential neo-epitopes that can elicit antidrug immune responses13,39 with serious clinical consequences.28 The ability to identify and mitigate potentially immune sequences in engineered proteins is therefore a high priority during the early-stage development of bioengineered therapeutic proteins.13
Animal models, the workhorses of preclinical toxicity assessment, have limited utility in predicting clinical immunogenicity of protein therapeutics. Differences between the human and animal immunogenicity responses, the lack of genetic diversity in animal models, and differences in dose results in animal studies are among limiting factors when assigning immunogenicity risk.40 The molecule VA described in this study was investigated in 2 animal models. Immune responses determined by enzyme-linked immunosorbent assay revealed no statistically significant difference between the responses to the WT-rFVIIa and VA.41 This finding was supported by the phase 2 clinical trial in which no immunogenicity risk for VA was detected. However, a lager cohort of patients with greater HLA diversity in phase 3 clinical studies resulted in ADA responses in 11.1% of the patients.
Here we have used computational tools and ex vivo assays using human cells that are fit for purpose to design and evaluate a strategy to deimmunize a neo-epitope introduced while reengineering the rFVIIa analog, VA. The peptide-HLA-DRB1 affinity had been previously demonstrated to be a good surrogate marker of immune responses in patients exhibiting clinical immunogenicity.9 We therefore exploited computational strategies to disrupt the foreign-peptide HLA binding for a broad repertoire of HLA-DRB1 variants while also incorporating in silico tools to ensure that the desired protein functionality is preserved.
Our previous studies with VA have shown that T-cell assays using cells from blood bank donors can be good predictors of clinical immunogenicity.9 The assay has the added advantage that the cells can be collected from donor cohorts defined by HLA typing allowing us to construct an ex vivo model of a population of interest. Using 2 independent T-cell assays, a thymidine uptake proliferation assay and an IL-2 ELISpot, we demonstrate that deimmunized variants of VA do not lead to proliferation of T cells from any donor. Conversely, cells from ∼12% of donors responded to peptides with the VA sequence. The deimmunized variants of VA thus had immunogenicity profiles comparable to the marketed WT FVIIa product. VA and the marketed WT-rFVIIa have been subjected to a head-to-head comparison in a clinical study28 and the ex vivo assays described here9 with comparable outcomes (Figure 6B). The absence of the responses in cells from donors with a broad distribution of HLA-DRB1 variants strongly suggests that the computationally designed VA variants may have a favorable risk profile with respect to immunogenicity.
Our computational strategy was to lower immunogenicity risk while retaining the desired supraphysiologic functionality encoded by mutations in the VA molecule. We synthesized, purified, and tested in a functional assay the original VA and its deimmunized variants, which were predicted to retained VA function, as well as a variant expected to lose function. The 2 deimmunized variants of VA (N301D, N301E) predicted to retain the supraphysiologic functionality exhibited kinetics comparable to VA. The variant, which was predicted to lose function (L300E), showed no activity in the functional TGA method. As shown in Figure 5B, the deimmunized analogs exhibit comparable activity to that of VA while simultaneously having a lower risk of immunogenicity comparable to the WT-rFVIIa (Figure 6B). Based on these proof-of-concept experiments, the RID algorithm described here has been demonstrated to be well suited to designing optimal therapeutic proteins that are coengineered for desired functional attributes and minimal immunogenicity risk. Nonetheless, it will be necessary to demonstrate in clinical studies that the deimmunized molecule is indeed less immunogenic.
The software (RID) is available upon request.
The full-text version of this article contains a data supplement.
Acknowledgments
Research conducted in the laboratory of Z.E.S. is funded by US Food and Drug Administration (FDA) intramural grants from the Chief Scientist’s Challenge Grant and the Critical Path Initiative. This project was supported in part by an appointment of J. McGill to the Research Participation Program at the Office of Tissues and Advanced Therapies, Center for Biologics Evaluation and Research, US Food and Drug Administration, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the US Department of Energy and the FDA.
The findings and conclusions in this article have not been formally disseminated by the Food and Drug Administration and should not be construed as representing any Agency determination or policy.
Authorship
Contribution: W.J. and Z.E.S. designed the research; W.J., J. McGill, H.A.D.L., S.S., G.B., C.B., E.C., M.H.F., K.I.J., and A.K. performed the research; W.J., J. McGill, H.A.D.L., S.S., G.B., A.K., J. Marcotrigiano, M.V.O., and Z.E.S. analyzed the data; and W.J., and Z.E.S. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Zuben E. Sauna, Center for Biologics Evaluation and Research, US Food and Drug Administration, 10903 New Hampshire Ave, Building 52, Room 4120, Silver Spring, MD 20993; e-mail: zuben.sauna@fda.hhs.gov.

