

Key Points
Hematopoietic cell–targeted antibody-drug conjugate preconditioning is highly effective for platelet gene therapy in hemophilia A mice.
Platelet-specific FVIII gene therapy can effectively prevent a needle-induced knee joint injury in hemophilia A mice.

Abstract
Gene therapy offers the potential to cure hemophilia A (HA). We have shown that hematopoietic stem cell (HSC)–based platelet-specific factor VIII (FVIII) (2bF8) gene therapy can produce therapeutic protein and induce antigen-specific immune tolerance in HA mice, even in the presence of inhibitory antibodies. For HSC-based gene therapy, traditional preconditioning using cytotoxic chemotherapy or total body irradiation (TBI) has been required. The potential toxicity associated with TBI or chemotherapy is a deterrent that may prevent patients with HA, a nonmalignant disease, from agreeing to such a protocol. Here, we describe targeted nongenotoxic preconditioning for 2bF8 gene therapy utilizing a hematopoietic cell–specific antibody-drug conjugate (ADC), which consists of saporin conjugated to CD45.2- and CD117-targeting antibodies. We found that a combination of CD45.2- and CD117-targeting ADC preconditioning was effective for engrafting 2bF8-transduced HSCs and was favorable for platelet lineage reconstitution. Two thirds of HA mice that received 2bF8 lentivirus-transduced HSCs under (CD45.2+CD117)-targeting ADC conditioning maintained sustained therapeutic levels of platelet FVIII expression. When CD8-targeting ADC was supplemented, chimerism and platelet FVIII expression were significantly increased, with long-term sustained platelet FVIII expression in all primary and secondary recipients. Importantly, immune tolerance was induced and hemostasis was restored in a tail-bleeding test, and joint bleeding also was effectively prevented in a needle-induced knee joint injury model in HA mice after 2bF8 gene therapy. In summary, we show for the first time efficient engraftment of gene-modified HSCs without genotoxic conditioning. The combined cocktail ADC-mediated hematopoietic cell–targeted nongenotoxic preconditioning that we developed is highly effective and favorable for platelet-specific gene therapy in HA mice.
Introduction
Our previous studies have demonstrated that targeting factor VIII (FVIII) expression to platelets through hematopoietic stem cell (HSC)–based platelet-specific (2bF8) gene therapy restores hemostasis and induces immune tolerance in hemophilia A (HA) mice.1,,-4 However, in our protocol, sufficient bone marrow (BM) preconditioning was found to be essential to create a permissive environment to enable engraftment of the 2bF8 genetically modified HSCs. Prior preconditioning regimens used in our platelet gene therapy protocols involve total body irradiation (TBI) and/or chemotherapy using cytotoxic drugs, which are nontargeted and genotoxic, carrying potential risks for tissue damage, cytopenias, and secondary malignancy.5,,-8 The potential toxicities associated with this preconditioning present a barrier that may lessen the willingness of patients with HA to accept HSC-based platelet-targeted gene therapy. Thus, developing a protocol with targeted and less toxic preconditioning is desired to increase the safety and acceptance of such HSC-based gene therapy.
Recently, several novel proof-of-concept antibody-mediated preconditioning methods have been developed for BM transplantation (BMT) and HSC transplantation (HSCT). Initially, an antagonistic CD117 antibody that blocks stem cell growth factor receptor c-kit function was shown to enable efficient engraftment of donor cells in various immunocompromised disease models through depletion of host HSCs.9,-11 However, utilizing anti-CD117 antibody alone as a preconditioning for BMT/HSCT was insufficient in wild-type (WT) immunocompetent mice, yet a combination of anti-CD117 antibody with low-dose TBI or a CD47 antibody was effective.12,13 Subsequently, CD45 (leukocyte common antigen) or CD117 antibody-drug conjugated to protein synthesis toxin saporin (SAP), a plant ribosome-inactivating protein that halts protein synthesis,14,15 was shown to enable engraftment in immunocompetent WT mice.16,-18 SAP lacks a general cell entry domain and is nontoxic unless conjugated to a targeting antibody or ligand capable of receptor-mediated internalization.14,15 SAP and other protein-based immunotoxins have been widely explored in cancer therapy.15,19,,,,,,-26 Thus, utilizing a CD45-targeting antibody-drug conjugate (CD45-ADC) and/or a CD117-ADC could be a promising safe targeted nongenotoxic preconditioning regimen for BMT/HSCT; however, this combination has only been tested with syngeneic or allogeneic donor BM cells, and utility with transduced gene-modified cells is unknown.
In the current study, we evaluated antibody-drug conjugate (ADC)-based conditioning with platelet-directed HSC-based FVIII gene therapy in HA mice. We explored whether hematopoietic cell–targeted ADC preconditioning is effective for engraftments that are genetically manipulated by 2bF8 lentivirus (2bF8LV) and whether sustained therapeutic platelet FVIII expression is attainable in platelet-specific gene therapy utilizing ADC-based preconditioning.
Materials and methods
Antibodies and reagents
Details about the antibodies and reagents used in this study are provided in supplemental Materials and methods.
Mice
HA (FVIIInull) mice with the CD45.1 or CD45.2 congenic marker were established by the Shi laboratory by crossing C57BL/6 (B6)/129S mixed-background FVIIInull mice27 onto the CD45.1/B6 or CD45.2/B6 background. WT B6 mice and CAG-GFP–transgenic (GFPTg) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Isoflurane or ketamine was used for anesthesia. Animal studies were performed according to a protocol approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin.
ADC preconditioning, HSC transduction, and HSCT
ADCs were prepared by combining biotinylated antibody with streptavidin-SAP, as previously described.16,-18 The combination of CD45.2-ADC (3 mg/kg) plus CD117-ADC (0.5 mg/kg), with or without additional CD4-ADC (0.5 mg/kg) or CD8-ADC (0.5 mg/kg), was administered IV to 5- to 6-week-old FVIIInull/CD45.2 recipients 2 days before transplantation. Donors were congenic CD45.1/FVIIInull mice or CD45.2/GFPTg mice. BM cells were collected from donor mice, and Sca-1+ cells were isolated as previously described.1,28,29 CD45.1/FVIIInull Sca-1+ cells were transduced with 2bF8LV, as previously described,1,,-4 and 2.5 × 106 cells per mouse were transplanted into CD45.2/FVIIInull recipients preconditioned with ADCs. Some animals preconditioned with (CD45.2+CD117)-ADC were transplanted with an additional 1 × 107 Sca-1–depleted untransduced leukocytes to investigate the impact of donor hematopoietic cell number on BM reconstitution. The prior optimal sublethal 6.6-Gy TBI4 was used as a control regimen in parallel. Recipients were analyzed beginning at 3 weeks after transplantation.
Peripheral blood analysis
PCR and qPCR
The viability of 2bF8LV-transduced cells was examined by polymerase chain reaction (PCR) and quantitative real-time PCR (qPCR), as previously described.31 Details are provided in supplemental Materials and methods.
FVIII activity assay
Phenotypic correction assessment
The bleeding phenotype in recipients was assessed by a tail-bleeding test and a joint injury model. We used a 6-hour tail-bleeding test to grade the phenotypic correction in 2bF8LV-transduced recipients, as previously described.3,4,29,33 For the joint injury model, we used a needle-induced knee joint injury, as reported.34,,-37 Details are provided in supplemental Materials and methods.
FVIII immune responses study
After 24 weeks of gene therapy, animals were challenged with recombinant human FVIII (rhF8) at a dose of 100 U/kg or 200 U/kg weekly for 4 weeks by IV administration. The titers of inhibitory antibodies (inhibitors) before and after rhF8 immunization were determined by Bethesda assay, as previously described.30 Details are provided in supplemental Materials and methods.
Statistical analysis
Data are presented as the mean ± standard deviation. Details of the statistical comparisons of experimental groups are provided in supplemental Materials and methods.
Results
ADC-mediated preconditioning enables HA mice to engraft HSCs that are genetically modified by 2bF8LV
To examine whether ADC conditioning is capable of creating a niche for engrafting 2bF8LV-transduced HSCs, we monitored peripheral blood counts and leukocyte chimerism after transplantation of 2bF8LV-transduced HSCs. Pilot experiments demonstrated that after using 3 mg/kg CD45.2-ADC or 1.5 mg/kg CD117-ADC preconditioning alone, only 20% of animals had sustained levels of platelet FVIII expression after 2bF8 gene therapy (data not shown). Thus, for subsequent experiments, we combined 3 mg/kg CD45-ADC and 0.5 mg/kg CD117-ADC into a single preconditioning regimen for 2bF8 gene therapy (Figure 1A). We found that at 3 weeks after HSCT, peripheral blood counts in the ADC-mediated conditioning groups were significantly higher than in the TBI group (Figure 1B-D), suggesting that using ADC-mediated preconditioning for platelet gene therapy can reduce the risk of cytopenia.

The influence of ADC-mediated conditioning on peripheral blood recovery and leukocyte chimerism in HA mice after 2bF8 lentiviral gene delivery to HSCs. CD45.2/FVIIInull mice were preconditioned with CD45.2 and CD117 antibodies conjugated with SAP and transplanted with Sca-1+ cells that were isolated from CD45.1/FVIIInull donors and transduced with 2bF8LV. Additional untransduced Sca-1–depleted cells were transplanted into some recipients. TBI (6.6-Gy) preconditioning was used as a control regimen in parallel. After ≥3 weeks of BM reconstitution, blood samples were collected for whole-blood count and flow cytometry analysis of leukocyte chimerism. (A) Schematic diagram of experimental design for 2bF8 gene therapy using (CD45.2+CD117)-ADC preconditioning. Sca-1+ cells isolated from CD45.1/FVIIInull donors were transduced with 2bF8LV and transplanted into CD45.2/FVIIInull recipients preconditioned with (CD45.2+CD117)-ADC. Some recipients were cotransplanted with untransduced Sca-1–depleted hematopoietic cells. (B) Leukocyte counts. (C) Platelet counts. (D) Hemoglobin levels. (E) Donor-derived leukocyte chimerism. (F) Percentage of animals with >15% donor-derived leukocytes at 20 weeks after transplantation. (G) Percentage of donor-derived leukocytes at 20 weeks after transplantation. These results show that (CD45.2+CD117)-ADC conditioning does not result in prolonged cytopenias in HA mice and enables engraftment of 2bF8LV-transduced HSCs after platelet-directed gene therapy. *P < .05, **P < .01, ***P < .001, ****P < .0001, ADC vs TBI, 2-way analysis of variance. Bio, biotin; SA, streptavidin.
The influence of ADC-mediated conditioning on peripheral blood recovery and leukocyte chimerism in HA mice after 2bF8 lentiviral gene delivery to HSCs. CD45.2/FVIIInull mice were preconditioned with CD45.2 and CD117 antibodies conjugated with SAP and transplanted with Sca-1+ cells that were isolated from CD45.1/FVIIInull donors and transduced with 2bF8LV. Additional untransduced Sca-1–depleted cells were transplanted into some recipients. TBI (6.6-Gy) preconditioning was used as a control regimen in parallel. After ≥3 weeks of BM reconstitution, blood samples were collected for whole-blood count and flow cytometry analysis of leukocyte chimerism. (A) Schematic diagram of experimental design for 2bF8 gene therapy using (CD45.2+CD117)-ADC preconditioning. Sca-1+ cells isolated from CD45.1/FVIIInull donors were transduced with 2bF8LV and transplanted into CD45.2/FVIIInull recipients preconditioned with (CD45.2+CD117)-ADC. Some recipients were cotransplanted with untransduced Sca-1–depleted hematopoietic cells. (B) Leukocyte counts. (C) Platelet counts. (D) Hemoglobin levels. (E) Donor-derived leukocyte chimerism. (F) Percentage of animals with >15% donor-derived leukocytes at 20 weeks after transplantation. (G) Percentage of donor-derived leukocytes at 20 weeks after transplantation. These results show that (CD45.2+CD117)-ADC conditioning does not result in prolonged cytopenias in HA mice and enables engraftment of 2bF8LV-transduced HSCs after platelet-directed gene therapy. *P < .05, **P < .01, ***P < .001, ****P < .0001, ADC vs TBI, 2-way analysis of variance. Bio, biotin; SA, streptavidin.
Flow cytometry analysis at 3 weeks after HSCT showed 2.4% ± 2.2% and 3.5% ± 2.7% donor-derived cell chimerism in the (CD45.2+CD117)-ADC preconditioned groups transplanted with 2.5 × 106 2bF8LV-transduced FVIIInull/CD45.1 Sca-1+ cells or 2.5 × 106 2bF8LV-transduced Sca-1+ cells plus 10 × 106 Sca-1–depleted untransduced FVIIInull/CD45.1 leukocytes, respectively; these values were significantly lower than in the TBI group (73.9% ± 6.1%) (Figure 1E). However, by 5 months post-HSCT, engraftment in the ADC-preconditioned groups increased to 41.5% ± 26.6% and 34.6% ± 33.8%, respectively, whereas the chimerism in the TBI control group remained stable, and there were no statistically significant differences among the groups. By 20 weeks post-HSCT, all transplant recipients under (CD45.2+CD117)-ADC conditioning had >15% donor-derived leukocytes (Figure 1F-G). Under (CD45.2+CD117)-ADC conditioning, after transplantation, donor-derived subset chimerism gradually increased, and endogenous cells decreased with time (supplemental Figure 1). Multilineage donor engraftment was observed; however, donor-derived granulocyte reconstitution was more rapid than CD4, CD8, and B cell reconstitution between 3 and 15 weeks (supplemental Figure 1A). Together, these data show that long-term engraftment can be achieved utilizing a preconditioning regimen combination CD45.2-ADC and CD117-ADC.
ADC-mediated conditioning enables sustained platelet FVIII expression in HA mice after 2bF8 gene therapy
To validate whether (CD45.2+CD117)-ADC conditioning is effective in maintaining sustained neoprotein platelet FVIII expression in FVIIInull mice after 2bF8 lentiviral gene delivery to HSCs, we monitored 2bF8 proviral DNA and platelet FVIII expression in transduced recipients. The 2bF8 expression cassette was detected in all 2bF8LV-transduced recipients under (CD45.2+CD117)-ADC or TBI conditioning by PCR, demonstrating that 2bF8LV-transduced HSCs were viable under ADC-mediated preconditioning. Representative results are shown in Figure 2A. qPCR analysis showed that, at 4 weeks after transplantation, the average copy numbers of 2bF8 proviral DNA per cell in the (CD45.2+CD117)-ADC groups transplanted with 2bF8LV-transduced Sca-1+ cells or 2bF8LV-transduced Sca-1+ cells plus depleted cells were significantly lower than in the TBI control group. The copy number of 2bF8 transgenes per cell increased with time after transplantation in the ADC groups and reached a similar level as in the TBI group by 16 weeks post-BMT (Figure 2B).
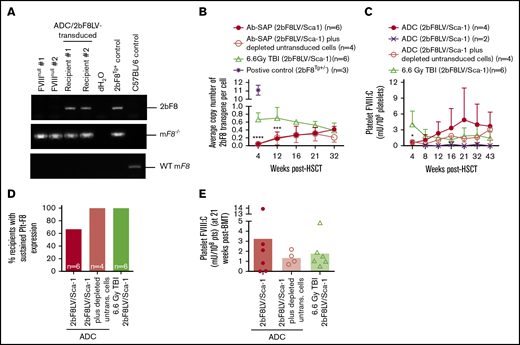
Platelet FVIII expression in HA mice after 2bF8 lentiviral gene delivery to HSCs under ADC-mediated preconditioning. Blood samples were collected at various time points from 2bF8LV-transduced recipients under (CD45.2+CD117)-ADC–mediated preconditioning or TBI. Leukocytes and platelets were isolated. DNA was purified from leukocytes for PCR and qPCR analysis of proviral DNA. Platelet FVIII expression levels were determined by a chromogenic functional activity assay on platelet lysates. (A) PCR detection of the 2bF8 transgene expression cassette. 2bF8 proviral DNA was detected in leukocytes in all 2bF8LV-transduced recipients. Representative results are shown. FVIIInull and WT mice were used as controls. (B) The average copy number of 2bF8 proviral DNA per cell in leukocytes from 2bF8LV-transduced recipients, as determined by qPCR. Heterozygous 2bF8-transgenic mice (2bF8Tg+/−) that were generated by our group via embryonic stem cell (ES)–mediated transgenesis with a known copy number (11 copies per cell) were used as a positive control. (C) The platelet FVIII expression levels in platelets from 2bF8LV-transduced recipients. Two of 6 recipients under (CD45.2+CD117)-ADC conditioning that received 2bF8LV-transduced Sca-1 cells failed to achieve platelet FVIII expression. All other recipients had sustained platelet FVIII expression. (D) Percentage of recipients with sustained platelet FVIII expression. (E) Platelet FVIII expression levels at 21 weeks after transplantation. These results demonstrate that sustained platelet FVIII expression can be achieved in HA mice after 2bF8 gene therapy with ADC-mediated preconditioning. *P < .05, ***P < .001, ****P < .0001, ADC vs TBI, 2-way analysis of variance.
Platelet FVIII expression in HA mice after 2bF8 lentiviral gene delivery to HSCs under ADC-mediated preconditioning. Blood samples were collected at various time points from 2bF8LV-transduced recipients under (CD45.2+CD117)-ADC–mediated preconditioning or TBI. Leukocytes and platelets were isolated. DNA was purified from leukocytes for PCR and qPCR analysis of proviral DNA. Platelet FVIII expression levels were determined by a chromogenic functional activity assay on platelet lysates. (A) PCR detection of the 2bF8 transgene expression cassette. 2bF8 proviral DNA was detected in leukocytes in all 2bF8LV-transduced recipients. Representative results are shown. FVIIInull and WT mice were used as controls. (B) The average copy number of 2bF8 proviral DNA per cell in leukocytes from 2bF8LV-transduced recipients, as determined by qPCR. Heterozygous 2bF8-transgenic mice (2bF8Tg+/−) that were generated by our group via embryonic stem cell (ES)–mediated transgenesis with a known copy number (11 copies per cell) were used as a positive control. (C) The platelet FVIII expression levels in platelets from 2bF8LV-transduced recipients. Two of 6 recipients under (CD45.2+CD117)-ADC conditioning that received 2bF8LV-transduced Sca-1 cells failed to achieve platelet FVIII expression. All other recipients had sustained platelet FVIII expression. (D) Percentage of recipients with sustained platelet FVIII expression. (E) Platelet FVIII expression levels at 21 weeks after transplantation. These results demonstrate that sustained platelet FVIII expression can be achieved in HA mice after 2bF8 gene therapy with ADC-mediated preconditioning. *P < .05, ***P < .001, ****P < .0001, ADC vs TBI, 2-way analysis of variance.
All recipients in the (CD45.2+CD117)-ADC group transplanted with 2bF8LV-transduced Sca-1+ cells plus depleted cells had sustained platelet FVIII expression; the levels of platelet FVIII were not statistically significantly different from those in the TBI group at various time points throughout the study course (Figure 2C). In the (CD45.2+CD117)-ADC group transplanted with only 2bF8LV-transduced Sca-1+ cells, 4 of 6 recipients had sustained platelet FVIII expression, and the other 2 did not (Figure 2C-E). Of those 4 recipients, platelet FVIII expression increased significantly from 0.48 ± 0.45 mU per 108 platelets at 4 weeks and reached a plateau level of 4.88 ± 6.05 mU per 108 platelets by 21 weeks after transplantation. This appears to be higher than the levels obtained in the TBI control group (1.80 ± 1.56 mU per 108 platelets) and in the ADC group transplanted with 2bF8LV-transduced Sca-1+ cells plus depleted cells (1.36 ± 0.66 mU per 108 platelets) at this time point, but there were no statistically significant differences among the groups (Figure 2C). No FVIII was detected in plasma (data not shown). Taken together, these data demonstrate that sustained platelet FVIII expression can be achieved in 2bF8 gene therapy using ADC-mediated preconditioning.
ADC-mediated conditioning is favorable to platelet lineage reconstitution in the early phase after HSCT
It was surprising that the ADC group exhibited such low leukocyte chimerism but decent levels of platelet FVIII expression. Because there is no congenic marker available to track platelet chimerism, we used GFP mice as donors to investigate platelet reconstitution under (CD45.2+CD117)-ADC conditioning. Freshly isolated Sca-1+ cells from GFPTg mice were transplanted into CD45.2/FVIIInull recipients preconditioned with ADC (Figure 3A). Interestingly, 86.7% ± 10.4% of platelets in recipients were derived from GFPTg donors by 3 weeks post-BMT (Figure 3B), but only 23.7% ± 13.1% of leukocytes were GFP+ (Figure 3C). Platelet chimerism increased to >90% by 7 weeks of BM reconstitution after HSCT. Leukocytes gradually increased with time, reaching 76.9% ± 18.1% by 19 weeks after HSCT. B cells and myeloid cells reconstituted faster than CD4+ and CD8+ T cells after HSCT under (CD45.2+CD117)-ADC conditioning (Figure 3D-G).
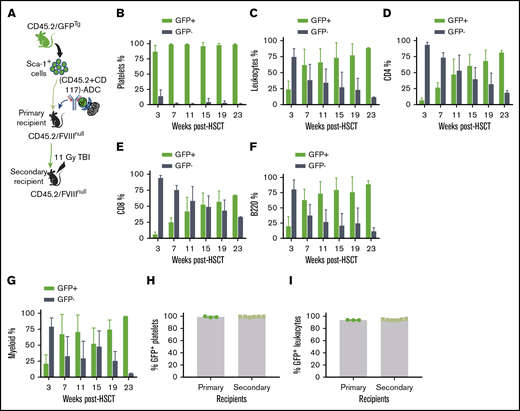
The influence of ADC-mediated preconditioning on blood cell lineage reconstitution after HSCT. Sca-1 cells were isolated from CD45.2/CAG-GFP–transgenic (CD45.2/GFPTg) mice and transplanted into CD45.2/FVIIInull mice under (CD45.2+CD117)-ADC preconditioning. Platelets were stained with CD41 antibody. Leukocytes were stained with CD45.2, CD4, CD8, and B220 antibodies, and donor-derived GFP+ chimerism was analyzed by flow cytometry at various time points after HSCT. (A) Schematic diagram of experimental design for GFPTg HSCT using (CD45.2+CD117)-ADC preconditioning. (B) Platelet chimerism. (C) Leukocyte chimerism. (D) CD4 T cell chimerism. (E) CD8 T cell chimerism. (F) B-cell chimerism. (G) Myeloid chimerism. (H) Sustained high levels of platelet chimerism in primary and secondary transplant recipients. (I) Sustained leukocyte chimerism in primary and secondary transplant recipients. These results demonstrate that (CD45.2+CD117)-ADC preconditioning is favorable for platelet lineage reconstitution after HSCT.
The influence of ADC-mediated preconditioning on blood cell lineage reconstitution after HSCT. Sca-1 cells were isolated from CD45.2/CAG-GFP–transgenic (CD45.2/GFPTg) mice and transplanted into CD45.2/FVIIInull mice under (CD45.2+CD117)-ADC preconditioning. Platelets were stained with CD41 antibody. Leukocytes were stained with CD45.2, CD4, CD8, and B220 antibodies, and donor-derived GFP+ chimerism was analyzed by flow cytometry at various time points after HSCT. (A) Schematic diagram of experimental design for GFPTg HSCT using (CD45.2+CD117)-ADC preconditioning. (B) Platelet chimerism. (C) Leukocyte chimerism. (D) CD4 T cell chimerism. (E) CD8 T cell chimerism. (F) B-cell chimerism. (G) Myeloid chimerism. (H) Sustained high levels of platelet chimerism in primary and secondary transplant recipients. (I) Sustained leukocyte chimerism in primary and secondary transplant recipients. These results demonstrate that (CD45.2+CD117)-ADC preconditioning is favorable for platelet lineage reconstitution after HSCT.
To investigate whether high chimerism in platelets under ADC-mediated conditioning is due to BM bias to platelet lineage reconstitution or truly high chimerism of HSCs in BM, sequential transplantation was carried out at 56 weeks after HSCT by transplanting BM cells from primary recipients into secondary recipients under a lethal 11-Gy TBI. After 3 weeks of BM reconstitution, animals were analyzed by flow cytometry. Platelet chimerism was maintained at 99.2% ± 0.2%, which was not significantly different compared with that in primary recipients (Figure 3H). Similarly, the chimerism in leukocytes in the secondary recipients was similar to the primary recipients (Figure 3I). The engraftment was stable in the secondary recipients (9 months post-BMT). These results suggest that high platelet chimerism in recipients under ADC-mediated conditioning was indeed from high HSC chimerism. Thus, our data indicate that ADC conditioning is favorable to platelet lineage reconstitution in the early phase after HSCT, suggesting that ADC conditioning is especially beneficial in our platelet-targeted gene therapy.
The addition of CD8-ADC enhances the efficacy of (CD45.2+CD117)-ADC conditioning in platelet gene therapy
Because 2 of the 6 FVIIInull recipients preconditioned with (CD45.2+CD117)-ADC and transplanted with 2bF8LV-transduced Sca-1+ cells did not have sustained platelet FVIII expression, we tested whether the addition of CD4-ADC or CD8-ADC (Figure 4A) would improve the efficacy. As shown in Figure 4B, at 3 weeks after HSCT, total leukocyte chimerism in the group with additional CD4-ADC and CD8-ADC was 23.6% ± 18.7% and 23.0% ± 7.9%, respectively, which was 10-fold greater than in the (CD45.2+CD117)-ADC group. The chimerism rapidly increased with time after HSCT, reaching 75.9% ± 14.6% and 78.1% ± 7.7% by 20 weeks (Figure 4C) in the (CD45.2+CD117+CD4)-ADC and (CD45.2+CD117+CD8)-ADC groups, respectively; these values were significantly higher than in the (CD45.2+CD117)-ADC group at the same time point as shown in Figure 1E (41.5 ± 26.6%) and at an earlier time point (supplemental Figure 2).

The impact of the addition of CD4-ADC or CD8-ADC on the efficacy of (CD45.2+CD117)-ADC preconditioning in platelet-specific FVIII gene therapy of HA mice. CD45.2/FVIIInull mice were preconditioned with (CD45.2+CD117)-ADC plus CD8-ADC or CD4-ADC and received 2bF8LV-transduced CD45.1/FVIIInull Sca-1+ cells. TBI was used as a control preconditioning regimen in parallel. After ≥3 weeks of BM reconstitution, blood samples were collected from recipients post-HSCT at various time points for chimerism analysis, qPCR analysis, and platelet lysate FVIII activity assay. (A) Schematic diagram of experimental design for 2bF8 gene therapy using (CD45.2+CD117)-ADC plus CD4-ADC or CD8-ADC preconditioning. (B) Leukocyte chimerism analyzed by flow cytometry. (C) Percentage of donor-derived leukocytes at 20 weeks after HSCT. (D) Platelet FVIII expression levels determined by platelet lysate FVIII activity assay. (E) Platelet FVIII expression levels at 21 weeks after HSCT. (F) Average copy number of 2bF8 proviral DNA per cell from peripheral blood leukocytes, as determined by qPCR. Homozygous 2bF8-transgenic mice (LV17/18Tg+/+) that were generated by our group via 2bF8LV-mediated transgenesis with a known copy number (4 copies per cell) were used as a positive control. (G) Platelet FVIII expression in sequential BMT recipients under ADC-mediated preconditioning. Sixty weeks after HSCT, BM cells were collected from some primary (1°) recipients and transplanted into secondary (2°) recipients under the same conditioning regimen that donors received: (CD45.2+CD117+CD4)-ADC or (CD45.2+CD117+CD8)-ADC. Animals were analyzed monthly. The levels of platelet FVIII expression from the 1° recipients were averaged from the last 3 time points. The 2° recipients’ platelet FVIII levels were averaged from the 3 time points after sequential transplantation. These results demonstrate that the addition of CD4-ADC or CD8-ADC can enhance the efficacy of (CD45.2+CD117)-ADC preconditioning in platelet-specific FVIII gene therapy in HA mice.
The impact of the addition of CD4-ADC or CD8-ADC on the efficacy of (CD45.2+CD117)-ADC preconditioning in platelet-specific FVIII gene therapy of HA mice. CD45.2/FVIIInull mice were preconditioned with (CD45.2+CD117)-ADC plus CD8-ADC or CD4-ADC and received 2bF8LV-transduced CD45.1/FVIIInull Sca-1+ cells. TBI was used as a control preconditioning regimen in parallel. After ≥3 weeks of BM reconstitution, blood samples were collected from recipients post-HSCT at various time points for chimerism analysis, qPCR analysis, and platelet lysate FVIII activity assay. (A) Schematic diagram of experimental design for 2bF8 gene therapy using (CD45.2+CD117)-ADC plus CD4-ADC or CD8-ADC preconditioning. (B) Leukocyte chimerism analyzed by flow cytometry. (C) Percentage of donor-derived leukocytes at 20 weeks after HSCT. (D) Platelet FVIII expression levels determined by platelet lysate FVIII activity assay. (E) Platelet FVIII expression levels at 21 weeks after HSCT. (F) Average copy number of 2bF8 proviral DNA per cell from peripheral blood leukocytes, as determined by qPCR. Homozygous 2bF8-transgenic mice (LV17/18Tg+/+) that were generated by our group via 2bF8LV-mediated transgenesis with a known copy number (4 copies per cell) were used as a positive control. (G) Platelet FVIII expression in sequential BMT recipients under ADC-mediated preconditioning. Sixty weeks after HSCT, BM cells were collected from some primary (1°) recipients and transplanted into secondary (2°) recipients under the same conditioning regimen that donors received: (CD45.2+CD117+CD4)-ADC or (CD45.2+CD117+CD8)-ADC. Animals were analyzed monthly. The levels of platelet FVIII expression from the 1° recipients were averaged from the last 3 time points. The 2° recipients’ platelet FVIII levels were averaged from the 3 time points after sequential transplantation. These results demonstrate that the addition of CD4-ADC or CD8-ADC can enhance the efficacy of (CD45.2+CD117)-ADC preconditioning in platelet-specific FVIII gene therapy in HA mice.
Importantly, all 2bF8LV-transduced recipients had sustained platelet FVIII expression when conditioned with (CD45.2+CD117)-ADC plus CD4-ADC or CD8-ADC (Figure 4D-E). At 4 weeks after HSCT, the level of platelet FVIII in the (CD45.2+CD117+CD8)-ADC group was 4.23 ± 1.03 mU per 108 platelets, which was significantly higher than in the (CD45.2+CD117)-ADC group (0.32 ± 0.43 mU per 108 platelets). The level of platelet FVIII in the (CD45.2+CD117+CD4)-ADC group was 2.35 ± 1.05 mU per 108 platelets at 4 weeks after HSCT. At 21 weeks after gene therapy, the levels of platelet FVIII in the (CD45.2+CD117+CD8)-ADC and (CD45.2+CD117+CD4)-ADC groups were 4.89 ± 2.16 mU per 108 platelets and 8.57 ± 6.39 mU per 108 platelets, respectively. qPCR analysis at 4 weeks showed an average of 0.36 ± 0.07 and 0.25 ± 0.15 copies per cell (Figure 4F) of 2bF8 provirus DNA in the (CD45.2+CD117+CD4)-ADC and (CD45.2+CD117+CD8)-ADC groups, respectively; these values were significantly higher than that obtained from the (CD45.2+CD117)-ADC group (Figure 2B) at the same time point. The average copy number of 2bF8 provirus DNA per cell increased significantly at 8 weeks and then remained stable throughout the study course (Figure 4F).
To ensure that long-term expression of platelet FVIII is achievable under ADC conditioning, we performed sequential BMT. Sixty weeks after receiving 2bF8LV-transduced Sca-1 cells, BM cells from primary recipients were transplanted into secondary recipients under the same conditioning regimen that donors received [(CD45.2+CD117+CD4)-ADC or (CD45.2+CD117+CD8)-ADC]. After BM reconstitution, animals were analyzed monthly. One sequential transplantation using (CD45.2+CD117+CD4)-ADC preconditioning did not retain sustained platelet FVIII expression (data not shown), but all recipients under (CD45.2+CD117+CD8)-ADC preconditioning had sustained platelet FVIII expression. Platelet FVIII levels in the secondary recipients were similar to their donors’ levels (primary recipients) (Figure 4G). These data demonstrate that 2bF8LV genetically manipulated long-term engrafting HSCs can be maintained and platelet FVIII expression can be sustained in HA mice under the (CD45.2+CD117+CD8)-ADC regimen.
Hemostatic efficacy is achieved in HA mice after platelet-specific FVIII gene therapy with ADC-mediated preconditioning
To validate the hemostatic effectiveness of 2bF8 gene therapy under ADC-mediated preconditioning, we used 2 in vivo injury models. The 6-hour tail-bleeding test was used to grade the phenotypic correction of the FVIIInull coagulation defect. The clotting time in 2bF8LV-transduced recipients under ADC conditioning with sustained platelet FVIII expression was 3.4 ± 1.5 hours, which was not significantly different from the TBI group (3.8 ± 1.3 hours) but was significantly shorter than in the FVIIInull control group (none were able to clot within 6 hours) (Figure 5A-B). The remaining hemoglobin levels in the ADC-preconditioned group was 75.9% ± 20.2%, which was significantly higher than in the FVIIInull control group (42.8% ± 12%). There was no statistical difference between the ADC-preconditioned group and the TBI group (76.5% ± 21.3%; n = 6) or the B6 WT control group (69.6% ± 3.4%). The 2 recipients in the ADC group that received 2bF8LV-transduced Sca-1+ cells with undetectable platelet FVIII had hemoglobin levels similar to FVIIInull controls (Figure 5C).
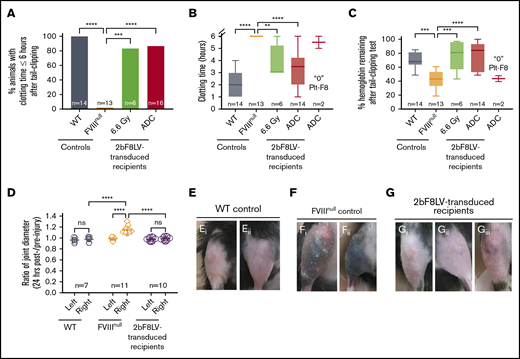
Phenotypic correction assessment of HA mice after platelet-specific gene therapy under ADC-mediated preconditioning. At least 4 months after 2bF8LV gene therapy, the bleeding phenotype in 2bF8LV-transduced recipients was assessed by a 6-hour tail-bleeding test and a needle-induced knee joint injury. For the tail-bleeding test, the tail tip was transected using a 1.6-mm-diameter template. Animals were monitored hourly, and clot time was recorded. Blood samples were collected before and after the test for blood counts. Hemoglobin level before the test was normalized to 100%. For the knee joint injury, a G30 × 1/2 needle was used to induce injury in the right knee, leaving the left knee unharmed as an intra-animal control. The diameter of the knee joint was measured using a digital micro caliper before and 24 hours after injury. The diameter of the knee joint before injury was defined as 1. WT and FVIIInull mice served as controls. (A) Percentage of animals whose tail bleeding stopped within 6 hours after tail tip transection. The statistical differences between the groups were analyzed using Fisher’s exact test. (B) Tail-bleeding time. Data from the 2 2bF8LV-transduced recipients in the (CD45.2+CD117)-ADC group that did not have sustained platelet FVIII expression are presented as a separate group. The statistical differences between the groups were analyzed using 1-way analysis of variance (ANOVA), followed by Tukey’s multiple-comparisons test. (C) Percentage of hemoglobin remaining after the tail-bleeding test. Data from the 2 2bF8LV-transduced recipients in the (CD45.2+CD117)-ADC group that did not have sustained platelet FVIII expression are presented as a separate group. The statistical differences between the groups were analyzed using 1-way ANOVA, followed by Tukey’s multiple-comparisons test. (D) Ratio of knee joint diameter measured at 24 hours postinjury/preinjury. The statistical differences between the groups were analyzed using 2-way ANOVA, followed by Tukey’s multiple-comparisons test. Representative images of the knee joint from WT control mice (E) and FVIIInull control mice (F) 24 hours after injury. (G) Representative images of the knee joint from 2bF8LV-transduced recipients with ADC preconditioning 24 hours after injury. These data demonstrate that effective phenotypic correction is attainable in HA mice after 2bF8 lentiviral gene delivery to HSCs under a nongenotoxic hematopoietic cell–targeted ADC preconditioning. **P < .01, ***P < .001, ****P < .0001. ns, not significant.
Phenotypic correction assessment of HA mice after platelet-specific gene therapy under ADC-mediated preconditioning. At least 4 months after 2bF8LV gene therapy, the bleeding phenotype in 2bF8LV-transduced recipients was assessed by a 6-hour tail-bleeding test and a needle-induced knee joint injury. For the tail-bleeding test, the tail tip was transected using a 1.6-mm-diameter template. Animals were monitored hourly, and clot time was recorded. Blood samples were collected before and after the test for blood counts. Hemoglobin level before the test was normalized to 100%. For the knee joint injury, a G30 × 1/2 needle was used to induce injury in the right knee, leaving the left knee unharmed as an intra-animal control. The diameter of the knee joint was measured using a digital micro caliper before and 24 hours after injury. The diameter of the knee joint before injury was defined as 1. WT and FVIIInull mice served as controls. (A) Percentage of animals whose tail bleeding stopped within 6 hours after tail tip transection. The statistical differences between the groups were analyzed using Fisher’s exact test. (B) Tail-bleeding time. Data from the 2 2bF8LV-transduced recipients in the (CD45.2+CD117)-ADC group that did not have sustained platelet FVIII expression are presented as a separate group. The statistical differences between the groups were analyzed using 1-way analysis of variance (ANOVA), followed by Tukey’s multiple-comparisons test. (C) Percentage of hemoglobin remaining after the tail-bleeding test. Data from the 2 2bF8LV-transduced recipients in the (CD45.2+CD117)-ADC group that did not have sustained platelet FVIII expression are presented as a separate group. The statistical differences between the groups were analyzed using 1-way ANOVA, followed by Tukey’s multiple-comparisons test. (D) Ratio of knee joint diameter measured at 24 hours postinjury/preinjury. The statistical differences between the groups were analyzed using 2-way ANOVA, followed by Tukey’s multiple-comparisons test. Representative images of the knee joint from WT control mice (E) and FVIIInull control mice (F) 24 hours after injury. (G) Representative images of the knee joint from 2bF8LV-transduced recipients with ADC preconditioning 24 hours after injury. These data demonstrate that effective phenotypic correction is attainable in HA mice after 2bF8 lentiviral gene delivery to HSCs under a nongenotoxic hematopoietic cell–targeted ADC preconditioning. **P < .01, ***P < .001, ****P < .0001. ns, not significant.
To evaluate the efficacy of platelet-specific FVIII gene therapy in preventing joint bleeding in HA mice, we produced a needle-induced knee joint injury in our 2bF8LV-transduced ADC-conditioned recipients. After a single needle puncture of the knee joint cavity, all FVIIInull control mice exhibited decreased activity and limped, and 9 of 11 mice exhibited visible bruising in the soft tissues around the injured knee joint at 24 hours after injury. In contrast, none of the 2bF8LV-transduced recipients (with a platelet FVIII level of 5.37 ± 4.49 mU per 108 platelets; range, 0.45-15.67 mU per 108 platelets) or WT mice exhibited decreased activity or limped after injury. Only 1 of the 10 recipients, which had the lowest platelet FVIII level (0.45 mU per 108 platelets), had a small bruise (Figure 5Giii) after needle-induced knee joint injury. When the diameters of the injured (right) knee joints were measured in FVIIInull mice, the ratio at 24 hours postinjury/preinjury joints was 1.143 ± 0.068 (n = 11); this was significantly higher compared with the ratio in the unharmed left joints (0.988 ± 0.027; P < .0001) (Figure 5D). It is notable that there are no significant differences in the ratios between the injured and unharmed intra-animal control joints in 2bF8LV-transduced recipients (0.988 ± 0.038 [right] and 0.976 ± 0.036 [left]), which are similar to the WT control (0.976 ± 0.049 [right] and 0.962 ± 0.042 [left]) (Figure 5D). Representative images of injured joints are shown in Figure 5E and G. These data demonstrate that platelet-specific gene therapy is effective in preventing joint bleeding in HA mice.
FVIII immune responses in platelet-specific FVIII gene therapy with ADC conditioning
Because regulatory T cells (Tregs) play an important role in modulating immune responses in our platelet-specific gene therapy model,4,28 we analyzed CD4+FoxP3+ Tregs at 11 weeks after 2bF8 gene therapy. The frequency of Tregs in 2bF8LV-transduced recipients in all groups was significantly higher than in the control CD45.1/FVIIInull group (Figure 6A). We further analyzed donor-derived Tregs (CD45.1+CD4+Foxp3+); their frequencies in 2bF8LV-transduced mice under various types of ADC conditioning or TBI were also significantly higher than in the control CD45.1/FVIIInull mice (Figure 6B).
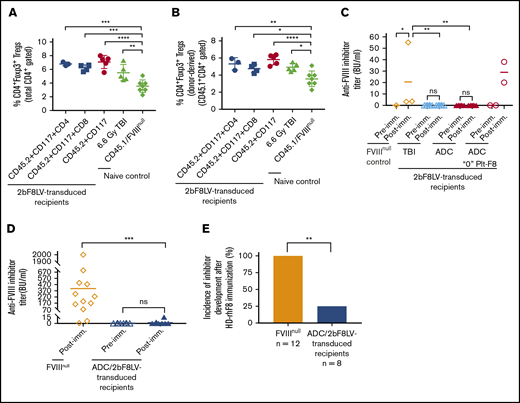
Immune responses in HA mice after platelet-specific gene therapy with hematopoietic cell–targeted ADC preconditioning. For Treg analysis, blood samples were collected from 2bF8LV-transduced recipients, and CD4+Foxp3+ Tregs were analyzed by flow cytometry. For immune response studies, ≥24 weeks after HSCT, recipients were immunized with rhF8 by IV infusion weekly for 4 weeks. One week after the last immunization, plasmas were collected, and inhibitor titers were determined by Bethesda assay. (A) Percentage of total Tregs in 2bF8LV-transduced recipients at 11 weeks after gene therapy. The statistical differences between the groups of transduced recipients and CD45.1/FVIIInull controls were analyzed using 1-way analysis of variance (ANOVA), followed by Dunnett’s multiple-comparisons test. (B) Percentage of donor-derived Tregs in 2bF8LV-transduced recipients at 11 weeks after gene therapy. The statistical differences between the groups of transduced recipients and CD45.1/FVIIInull controls were analyzed using 1-way ANOVA, followed by Dunnett’s multiple-comparisons test. (C) The anti-FVIII inhibitor titers in 2bF8LV-transduced recipients after challenge with rhF8 at a dose of 100 U/kg per week for 4 weeks. Data from the 2 2bF8LV-transduced recipients in the (CD45.2+CD117)-ADC group that did not have sustained platelet FVIII expression are presented as a separate group. The statistical differences between the groups were analyzed using 2-way ANOVA, followed by Tukey’s multiple-comparisons test. (D) The anti-FVIII inhibitor titers in 2bF8LV-transduced recipients after challenge with rhF8 at a dose of 200 U/kg per week for 4 weeks. The statistical differences between the groups were analyzed using the Mann-Whitney U test. (E) The incidence of inhibitor development in 2bF8LV-transduced recipients after challenge with rhF8 at a dose of 200 U/kg per week for 4 weeks. The statistical difference between the groups was analyzed using Fisher’s exact test. These data demonstrate that 2bF8 lentiviral gene delivery to HSCs under ADC-mediated preconditioning can suppress anti-FVIII immune responses. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Immune responses in HA mice after platelet-specific gene therapy with hematopoietic cell–targeted ADC preconditioning. For Treg analysis, blood samples were collected from 2bF8LV-transduced recipients, and CD4+Foxp3+ Tregs were analyzed by flow cytometry. For immune response studies, ≥24 weeks after HSCT, recipients were immunized with rhF8 by IV infusion weekly for 4 weeks. One week after the last immunization, plasmas were collected, and inhibitor titers were determined by Bethesda assay. (A) Percentage of total Tregs in 2bF8LV-transduced recipients at 11 weeks after gene therapy. The statistical differences between the groups of transduced recipients and CD45.1/FVIIInull controls were analyzed using 1-way analysis of variance (ANOVA), followed by Dunnett’s multiple-comparisons test. (B) Percentage of donor-derived Tregs in 2bF8LV-transduced recipients at 11 weeks after gene therapy. The statistical differences between the groups of transduced recipients and CD45.1/FVIIInull controls were analyzed using 1-way ANOVA, followed by Dunnett’s multiple-comparisons test. (C) The anti-FVIII inhibitor titers in 2bF8LV-transduced recipients after challenge with rhF8 at a dose of 100 U/kg per week for 4 weeks. Data from the 2 2bF8LV-transduced recipients in the (CD45.2+CD117)-ADC group that did not have sustained platelet FVIII expression are presented as a separate group. The statistical differences between the groups were analyzed using 2-way ANOVA, followed by Tukey’s multiple-comparisons test. (D) The anti-FVIII inhibitor titers in 2bF8LV-transduced recipients after challenge with rhF8 at a dose of 200 U/kg per week for 4 weeks. The statistical differences between the groups were analyzed using the Mann-Whitney U test. (E) The incidence of inhibitor development in 2bF8LV-transduced recipients after challenge with rhF8 at a dose of 200 U/kg per week for 4 weeks. The statistical difference between the groups was analyzed using Fisher’s exact test. These data demonstrate that 2bF8 lentiviral gene delivery to HSCs under ADC-mediated preconditioning can suppress anti-FVIII immune responses. *P < .05, **P < .01, ***P < .001, ****P < .0001.
We monitored anti-FVIII inhibitor titers in 2bF8LV-transduced recipients after transplantation. None of the recipients developed inhibitors after 2bF8 gene therapy with ADC-mediated preconditioning or TBI. To investigate whether immune tolerance was induced in 2bF8LV-transduced recipients, we immunized animals with 100 U/kg or 200 U/kg weekly for 4 weeks. With the 100-U/kg immunization protocol, none of the recipients (from the ADC or TBI group) developed inhibitors if they had sustained platelet FVIII expression. As a control, all 3 FVIIInull mice developed inhibitors with a level of 20.5 ± 29.9 BU/mL. The 2 recipients from the (CD45.2+CD117)-ADC group that received 2bF8LV-transduced Sca-1+ cells without sustained platelet FVIII expression did develop inhibitors with titers of 20 BU/mL and 38 BU/mL, respectively, after the rhF8 challenge (Figure 6C).
When a high-dose rhF8 (200-U/kg) immunization protocol that can elicit anti-FVIII immune responses, even in WT mice,38 was used, 2 of 8 2bF8LV-transduced recipients under ADC preconditioning developed inhibitors with titers of 3.5 BU/mL and 15 BU/mL. In contrast, all FVIIInull mice under the same immunization protocol developed inhibitors with an average titer of 406.4 ± 544.9 BU/mL (Figure 6D). The inhibitor titer (Figure 6D) and the incidence of inhibitor development (Figure 6E) in FVIIInull mice were significantly higher than in 2bF8LV-transduced recipients. Together, these data demonstrate that 2bF8LV gene delivery to HSCs under ADC conditioning is effective in suppressing anti-FVIII immune responses in FVIIInull mice.
Discussion
The central finding of this report is that platelet-directed HSC-based FVIII gene therapy is safe and effective for eliminating HA using nongenotoxic hematopoietic-targeted ADC conditioning. We show that ADC preconditioning enables long-term engraftment of 2bF8LV-transduced HSCs, resulting in sustained therapeutic levels of platelet FVIII expression, and ADC conditioning is favorable to platelet lineage reconstitution in the early phase after HSC transplantation. In the current study, we show for the first time efficient engraftment of in vitro lentivirus-mediated gene-engineered HSCs without genotoxic conditioning, as well as effective prevention of joint bleeding by platelet-targeted FVIII expression in HA mice.
Although HSCs are an attractive target for gene therapy of blood diseases, preconditioning is required to create space for engrafting the genetically manipulated cells. Potential toxicities associated with traditional preconditioning regimens (TBI or cytotoxic chemotherapy) could be a hurdle for patients to overcome, especially with nonmalignant diseases that otherwise do not require chemotherapy or irradiation as part of their standard treatment. Developing a protocol with targeted and nongenotoxic preconditioning is anticipated to increase acceptance of HSC-based gene therapy. In the current study, analysis of peripheral blood at 3 weeks after HSCT revealed normal blood counts in the ADC-preconditioned recipients, whereas cytopenias were observed in recipients of TBI. Our data demonstrate that ADC-based conditioning can minimize the cytopenic window associated with traditional irradiation-based conditioning and result in less severe potential risks associated with HSCT, highlighting the potential opportunity to perform this type of treatment in an outpatient setting.
It is worthwhile to point out that leukocyte chimerism was very low at the early time points after HSCT under ADC-mediated preconditioning, but platelet chimerism was fourfold greater than leukocyte chimerism, and platelet FVIII expression was comparable between the ADC and TBI groups. We reason that the (CD45.2+CD117)-ADC preconditioning regimen can preferentially target megakaryocytes and their progenitors because they may express higher levels of cell surface receptors CD45.2 and c-kit, leading to effective depletion of the platelet lineage. Thus, donor-derived cells have to compensate for platelet lineage reconstitution to maintain a balance of platelet numbers in the body, which leads to biased donor-derived platelets in recipients during the early stage of BM reconstitution. Indeed, it has been shown that CD117 is expressed on HSCs, as well as on megakaryocytes.39,,-42 In addition, a recent study demonstrated that different stages of megakaryocytes could have various levels of CD45 expression, with abundant expression on early progenitors and adult megakaryocytes.43 These findings support our notion that (CD45+CD117)-ADC preconditioning is especially beneficial for platelet lineage reconstitution and for our platelet-specific gene therapy by preferentially targeting the megakaryocyte lineage.
Our studies show that one third of 2bF8LV-transduced recipients that were preconditioned with (CD45.2+CD117)-ADC and received Sca-1+ cells did not have sustained therapeutic levels of platelet FVIII expression, although they had donor-derived leukocyte chimerism. When additional CD8-ADC was used, engraftment was significantly improved, and platelet FVIII expression was sustained in all recipients, including primary and secondary recipients. It is unclear why some recipients did not have sustained platelet FVIII expression under (CD45.2+CD117)-ADC conditioning, although leukocyte chimerism was obtained and proviral DNA was detectable by PCR and qPCR. We speculate that megakaryocyte precursors (eg, promegakaryoblasts) derived from 2bF8LV-transduced HSCs may be killed by recipient-derived residual CD8 T cells if platelet FVIII expression does not reach a threshold to induce immune tolerance. Because the αIIb promoter is active, but α-granules are absent, in megakaryoblasts, the neoprotein FVIII is expressed but not stored; therefore, megakaryoblasts might be targeted by killer cells if immune tolerance was not established. The relationship among the levels of platelet FVIII expression, preconditioning regimen, and immune tolerance is a subject warranting further investigation.
Studies from our group and other investigators have demonstrated that platelet-derived FVIII improves hemostasis in HA mice using various vessel injury models, including tail bleeding,2,-4,29,31,44 cuticular bleeding,45 electrolytic-induced femoral injury,2 FeCl3-induced carotid injury,45 and laser-induced cremaster injury45 models. However, the efficacy of platelet FVIII in preventing joint bleeding has not been evaluated. In our current study, we show that 2bF8 lentiviral gene delivery to HSCs under ADC-mediated preconditioning can effectively introduce platelet FVIII expression and rescue the hemophilic bleeding phenotype in a tail-bleeding test. Importantly, platelet-specific gene therapy can effectively prevent joint bleeding in a needle-induced knee joint injury in HA mice. Of note, the average amount of platelet FVIII in our transduced recipients subjected to joint injury corresponded to only 10.7% ± 8.9% (ranging from 1% to 31%) of FVIII activity in whole blood in normal WT mice. The clinical efficacy of such levels of platelet FVIII in preventing knee joint bleeding induced by needle puncture is remarkable. Our studies show that, if platelet FVIII expression is >1 mU per 108 platelets, the clinical efficacy is comparable to WT animals that have 100% plasma FVIII activity.
Our previous studies have demonstrated that lentivirus-mediated platelet-specific gene delivery to HSCs can induce immune tolerance to neoprotein in FVIII,3,4,29 FIX,33 and ovalbumin28 models. We have shown that the requirement of optimal preconditioning intensity for inducing immune tolerance is more stringent than is that for achieving sustained platelet FVIII expression in 2bF8 gene therapy in HA mice.4 Here, we show that phenotypic correction and immune tolerance are attainable in our HSC-based platelet-specific gene therapy using ADC-mediated nongenotoxic preconditioning. Our previous studies have demonstrated that antigen-specific Tregs are expanded after platelet-specific gene therapy, which is 1 of the mechanisms by which immune tolerance is induced.4,28 In the current study, we found that the frequencies of donor-derived and total Tregs from 2bF8LV-transduced recipients in groups under ADC-mediated preconditioning regimens were significantly higher than the frequency of naive controls, confirming that Tregs play an important role in immune tolerance induction in platelet-specific gene therapy.
In conclusion, we developed a cocktail of HSC-targeted ADCs as a preconditioning regimen that is highly effective and favorable for HSC-based platelet-directed gene therapy in HA mice. Because multiple similar clinical-grade HSC-targeted antibodies are in development and are being tested in clinical trials as conditioning agents for allogeneic HSCT, our studies highlight a promising rapidly translatable strategy for HSC-based gene therapy that could result in treatment of all genetic blood-based diseases and provide a platform for safe and sustained protein production for therapeutic purposes. This safe and effective treatment strategy could be especially meaningful for HA patients who are especially wary of standard preconditioning.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank H. Kazazian (University of Pennsylvania School of Medicine, Philadelphia, PA) for the FVIII-knockout mice and D. Derrick Rossi (Boston Children's Hospital, Boston, MA) for early guidance and initial ADCs to enable this work.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grant HL-102035 (Q.S.), as well as generous gifts from the Children’s Hospital of Wisconsin Foundation (Q.S.) and the Midwest Athletes Against Childhood Cancer Fund (Q.S.).
Authorship
Contribution: C.G. designed the study, performed experiments, and analyzed data; J.A.S. performed experiments and commented on the manuscript; F.X. and W.J. performed experiments and analyzed data; Y.C., A.S., and S.S. performed experiments; S.R. and H.W. contributed to the study design; A.C. contributed to the conception and the design of the study and commented on the manuscript; and Q.S. designed and conducted research, analyzed and interpreted data, and wrote the manuscript.
Conflict-of-interest disclosure: A.C. is an inventor on US patent applications (US 12/447,634; US 14/536,319; US 15/025,222; and US 15/148,837); and reports research funding from Rocket Pharmaceuticals, Inc.; equity ownership in and consultancy for Global Blood Therapeutics and Beam Therapeutics; equity ownership in, patents with, and royalties from Editas Medicines, Magenta Therapeutics, Forty Seven Inc., and Decibel Therapeutics; and employment with and equity ownership in GV Management Company. The remaining authors declare no competing financial interests.
Correspondence: Qizhen Shi, Department of Pediatrics, Medical College of Wisconsin, 8727 Watertown Plank Rd, Milwaukee, WI 53226; e-mail: qshi@versiti.org; and Agnieszka Czechowicz, Division of Stem Cell Transplantation and Regenerative Medicine, Department of Pediatrics, Stanford University School of Medicine, 300 Pasteur Dr, Grant Building Room H323, Stanford, CA 94304; e-mail: aneeshka@stanford.edu.

