

Key Points
GM-CSF is derived from both Th17/Tc17-positive and Th17/Tc17-negative donor lineages after bone marrow transplantation.
GM-CSF promotes the accumulation of alloantigen-presenting, migratory donor DCs in the gastrointestinal tract during GVHD.

Abstract
Granulocyte-macrophage colony-stimulating factor (GM-CSF) has recently emerged as an important pathogenic cytokine in acute graft-versus-host disease (GVHD), but the nature of the T-cell lineages secreting the cytokine and the mechanisms of action are less clear. Here we used interleukin 17A-fate reporter systems with transcriptional analysis and assays of alloantigen presentation to interrogate the origins of GM-CSF–secreting T cells and the effects of the cytokine on antigen-presenting cell (APC) function after experimental allogeneic stem cell transplantation (SCT). We demonstrated that although GM-CSF-secreting Th17 and non-Th17 cells expanded in the colon over time after SCT, the Th17 lineage expanded to represent 10% to 20% of the GM-CSF secreting T cells at this site by 4 weeks. Donor T-cell-derived GM-CSF expanded alloantigen-presenting donor dendritic cells (DCs) in the colon and lymph nodes. In the mesenteric lymph nodes, GM-CSF–dependent DCs primed donor T cells and amplified acute GVHD in the colon. We thus describe a feed-forward cascade whereby GM-CSF–secreting donor T cells accumulate and drive alloantigen presentation in the colon to amplify GVHD severity. GM-CSF inhibition may be a tractable clinical intervention to limit donor alloantigen presentation and GVHD in the lower gastrointestinal tract.
Introduction
Granulocyte-macrophage colony-stimulating factor (GM-CSF) is an inflammatory cytokine important in granulocyte and monocyte-macrophage expansion and differentiation.1 In allotransplantation, recipient macrophages are known to be regulatory2 and are important in limiting donor T-cell expansion by CD47-dependent pathways.3 Conversely, colony-stimulating factor 1 (CSF1)–dependent macrophages are potent inducers of fibrosis during chronic graft-versus-host disease (GVHD).4 Recently, pathogenic GM-CSF–secreting donor T cells have been described during GVHD.5,-7 These cells have been described as a distinct Th17-independent, but BATF (basic leucine zipper ATF-like transcription factor)-dependent, CD4+ T-cell lineage that preferentially induces GVHD in the colon.7 Reductions in GVHD in the absence of donor GM-CSF or its signaling was associated with impaired licensing of donor-derived phagocytes such that their production of inflammatory mediators including IL-1β and reactive oxygen species was attenuated.5
We and others have described pathogenic Th17 and Tc17 lineages after transplant that are RORγt (RAR-related orphan nuclear receptor γ t)-dependent pro-inflammatory T cells that produce cytokines of both Th1 and Th17 lineages, including the Th17 cytokine GM-CSF.8,,,-12 Th17/Tc17 cells are thus potent mediators of GVHD, including in the gastrointestinal (GI) tract.8,13 Importantly, these cells do not sustain interleukin 17 (IL-17) secretion over time and are best tracked with fate-reporter based systems.14 Acute GVHD is characterized by disruption to the GI tract and the activation and expansion of CD103+ donor dendritic cells (DCs) in the colon. These DCs present high levels of recipient alloantigen and migrate to the mesenteric nodes, where they expand and differentiate donor Th17/Th1 T cells and imprint gut-homing integrin receptors, leading to migration to the gut and fulminant GVHD.15,16
We have thus examined the lineage of GM-CSF–secreting donor T cells, using fate-reporting systems, and dissected the role of this cytokine on antigen presentation and T-cell priming in the GI tract during GVHD.
Methods
Mice
Female C57Bl/6 (B6 [H-2Db]) and BALB/c (H-2Dd) mice were purchased from the ARC (Animal Resources Center, WA, Australia). B6 background (H-2Db) IL-17Cre and Rosa26eYFP,14 TEa T-cell transgenic,15,17 GM-CSF−/−,18 those lacking the common β chain and murine-specific IL-3 β chain (lacking all signaling to GM-CSF, IL-3, and IL-5, βc−/−)19,20 and the B6.CD11cluc+ (CD11c-luciferase reporter)21 mice have been described. Mice were bred and housed in sterilized microisolator cages at either QIMR Berghofer or the Fred Hutchinson Cancer Research Center, and received acidified autoclaved water (pH 2.5) after transplantation. All animal experiments were approved by and performed in accordance with the QIMR Berghofer and Fred Hutchinson Cancer Research Center Animal Ethics Committees.
Bone marrow transplantation
Recipient BALB/c mice received 900 cGy total-body irradiation (137Cs source at 84.6 cGy/min) split over 2 doses (day −1). Recipients received 5 to 10 × 106 B6 T-cell depleted (TCD) BM cells with 0.2 to 0.25 × 106 purified T cells. T-cell depletion was performed by anti-CD4 (RL172.4), anti-CD8 (TIB211), and anti-Thy1.2 (HO-13-4) treatment, followed by rabbit complement. Cell suspensions contained less than 1% viable CD3+ T cells. T cells were purified by anti-CD19 (HB305), anti-B220 (RA36B2), anti-GR1, TER119, and anti-CD11b (TIB128) treatment followed by incubation with magnetic depletion via anti-rat immunoglobulin beads (Qiagen). Recipients of alloantigen-specific transgenic T cells received an intravenous injection on day 12 with 2 × 106 luciferase expressing TEa T cells. TEa (>98%; Vβ6+Vα2+) cells were purified by sorting (FACSAria; BD Biosciences).
Flow cytometry and monoclonal antibodies
Antibodies used in flow cytometry were purchased from either BioLegend (CD3 [145-2C11], CD4 [GK1.5], CD8α [53-6.7], CD90.2 [30-H12], IL-17A [TC11-18H10], GM-CSF [MP1-22E9], CD11c [N418], CD103 [M290], I-A/I-E [M5/114], CD11b [M1/70], CD45.1 [A20], CD45.2 [104], CCR7 [4B12]) or BD Biosciences (RORγt [Q31-378], BATF [S39-1060]). Biotinylated YAe (reactive to Ea peptide [peptide 52-68]) bound to I-Ab (the same complex recognized by TEa cells). Viability dyes (BioLegend) were included in all staining panels for dead cell exclusion. All samples were acquired on a BD LSR Fortessa (BD Biosciences), using BD FACSDiva (v8.0), and analyzed with FlowJo (v9.9.6).
Cell preparation, imaging, and cytokine analysis
Lymphocytes were isolated from mesenteric lymph nodes (mLNs) through mechanical disruption, or from the colon via murine Lamina Propria Dissociation Kit (Miltenyi Biotec), as per manufacturer instructions. Intracellular cytokine staining was performed as previously described,8,9,11 cells were cultured with phorbol 12-myristate 13-acetate (5 μg/mL) and ionomycin (50 μg/mL) for 4 hours with Brefeldin A (BioLegend), included in the final 3 hours of incubation. Cells were surface-labeled and processed for intracellular staining, cytokines were assessed via Cytofix/Cytoperm kit (BD Biosciences), and intranuclear staining was performed via fixation and permeabilization (eBioscience). TEaluc+ T-cell expansion was analyzed by luciferase signal intensity (Xenogen IVIS 100; Caliper Life Sciences). Light emission is presented as photons per second per square centimeter per steer radiant. Mice were injected with 500 µg d-Luciferin (subcutaneously; PerkinElmer) and imaged 5 minutes later.15
Gene expression analysis
Lymphocytes were isolated from the colon 4 weeks after allo-stem cell transplantation (SCT), using the murine Lamina Propria Dissociation Kit (Miltenyi Biotec), and target populations were sorted (>95% purity) from 6 mice. Total RNA was extracted with RNeasy Micro kits (Qiagen) and cDNA prepared with Maxima H Minus First Strand cDNA synthesis kits (Thermo Fisher). Quantitative polymerase chain reaction was performed using TaqMan gene expression assays (Applied Biosystems), and all measurements were run in parallel with, and normalized to, the housekeeping gene Hprt.
Statistical analysis
Statistical analyses were performed using GraphPad Prism versions 7 and 8 (GraphPad Software). Unpaired Student t tests (with Welch’s correction), where data were normally distributed, or Mann-Whitney U, where data were not normally distributed, were used to evaluate differences in cytokine expression and cell frequencies. Analysis of variance was used as indicated when comparisons were made between 3 or more groups. P < .05 was considered statistically significant.
Results
GM-CSF is derived from both Th17/Tc17-positive and Th17/Tc17-negative donor lineages after BMT
Although GM-CSF production has been attributed to a Th17-independent T-cell lineage after BMT,7 this is complicated by the fact that IL-17A is not a stable phenotypic marker of Th17 cells, and that Tc17 cells represent an important IL-17A–secreting lineage after BMT.8,11 We therefore examined GM-CSF secretion by T cells that were fate-reported for the Th17/Tc17 lineages, using IL-17CreRosa26eYFP donors (Figure 1A). GM-CSF secretion was clearly detectable from both Th17 and Tc17 cells, and although they are not the numerically dominant source of GM-CSF, the proportion of cells secreting GM-CSF was higher in Th17+ vs non-Th17 cells early after BMT in mLN and colon (Figure 1B). GM-CSF was also secreted by significantly higher proportions of Tc17 vs non-Tc17 cells at all points after BMT (Figure 1B). However, the frequencies and absolute numbers of GM-CSF+Tc17 in the colon and mLN were relatively low (Figure 1C-E). The frequency of GM-CSF+–secreting cells of the Th17 lineage increased over time in the colon, as did the number, representing 10% to 20% of the GM-CSF–secreting population by week 4 (Figure 1C-D). This increase in GM-CSF+Th17 cells in the colon over time was not seen in the mLN (Figure 1D), and the amount of GM-CSF secreted (as determined by mean fluorescence intensity) was similar in IL-17-positive and IL-17-negative fractions (data not shown). In contrast, the proportion of GM-CSF+–secreting cells of the Tc17 lineage was lower (approximately 5%), and neither the proportions nor absolute numbers increased over time (Figure 1C,E). Given that the numbers of both donor CD4+ and CD8+ T cells accumulate over time in the colon after BMT (Figure 1D-E), this suggests that both Th17 and non-Th17 GM-CSF+ cells preferentially accumulate at this site. GM-CSF expression by colonic CD4+ T cells has been reported to be BATF-dependent7 ; we therefore examined BATF expression more broadly after allo-SCT. Here we identified high levels of BATF expression in all donor T cells (CD4+ and CD8+), irrespective of RORγt or GM-CSF expression and the time assessed (Figure 1F-G). We observed a trend for increased BATF expression in RORγt+ cells, which because of reagent incompatibility could not be confirmed in YFP+ cells at the protein level. We therefore sorted YFP+ and YFP−CD4+ T cells from colon 4 weeks after allo-SCT, and performed quantitative polymerase chain reaction for BATF expression. Here we observed significantly higher Batf expression in CD4+YFP+ T cells compared with CD4+YFP− counterparts (Figure 1H).
![Th17/Tc17 are a significant source of GM-CSF in the GI tract after allo-SCT. Cytokine and transcription factor expression by CD4+YFP+ and CD8+YFP+ T cells was assessed in the colon and mLN 1, 2, and 4 weeks after allogeneic (B6.IL-17CreRosa26eYFP→Balb/c) transplant. (A) Representative flow cytometry plots gated on isotype controls demonstrating GM-CSF expression in YFP+ vs YFP− T cells 4 weeks after allo-SCT. (B) Frequency of GM-CSF expression in CD4+ and CD8+ T cells within YFP+ and YFP− compartments from colon and mLN at 1, 2, and 4 weeks after allo-SCT. (C) Frequency of YFP expression within CD4+GM-CSF+ and CD8+GM-CSF+ T cells from colon and mLN at 1, 2, and 4 weeks after allo-SCT. (D) Total number of CD4+ GM-CSF+YFP+, GM-CSF+YFP−, and total CD4+ T cells in colon and mLN at 1, 2, and 4 weeks after allo-SCT. (E) Total number of CD8+ GM-CSF+YFP+, GM-CSF+YFP−, and total CD8+ T cells in colon and mLN at 1, 2, and 4 weeks after allo-SCT. (A-E) The data are combined from 3 replicate experiments (n = 14-16 mice per group). (F) Representative histograms showing BATF expression in RORγt+ and RORγt− CD4+ and CD8+ T cell fractions in colon 2 and 4 weeks after allo-SCT. (G) Representative histograms showing BATF expression in GM-CSF+ and GM-CSF− colonic CD4+ T cells fractions 2 and 4 weeks after allo-SCT. (H) Batf relative gene expression in YFP+ and YFP− colonic CD4+ T cells 4 weeks after allo-SCT (n = 6 per group). Statistical analyses were performed by unpaired Student t test when comparing between 2 groups and 1-way analysis of variance when comparing over time (mean ± standard error of the mean [SEM]). *P < .05; **P < .01; ***P < .001; ****P < .0001.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/19/10.1182_bloodadvances.2019000053/3/m_advancesadv2019000053f1.png?Expires=1767851314&Signature=SOYV2On~cJIcT4jVpQkwqqMYjfM4g5gZz1LM1aryhu71F-1j1JUzDwbMSO4XjINSbsjdHye-TJLDhYVWOV0gRO2gtIjCTuQqal53nULcDGJiDgRx4umyC~MOZ3duGmXlDeEd-nBSH5ZVEn6ocUMjUR79xb7NaTVRJTzWOgQ1NkxGivqCbFCNiK2piS4hEToRZ9-oVbJudLn2zk1mqy5A~eYt4KzNnmWxJBWssWeNys~NRtLS~s5zqtwXu99npQul6mBlpwwFIcbHjE2~ObpT1P9MkjBVOxSlJ401UynntT0X~YMSeRf15IN-Z~9INk-EbG1Rh~ltISJlK1gM33FTaQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Th17/Tc17 are a significant source of GM-CSF in the GI tract after allo-SCT. Cytokine and transcription factor expression by CD4+YFP+ and CD8+YFP+ T cells was assessed in the colon and mLN 1, 2, and 4 weeks after allogeneic (B6.IL-17CreRosa26eYFP→Balb/c) transplant. (A) Representative flow cytometry plots gated on isotype controls demonstrating GM-CSF expression in YFP+ vs YFP− T cells 4 weeks after allo-SCT. (B) Frequency of GM-CSF expression in CD4+ and CD8+ T cells within YFP+ and YFP− compartments from colon and mLN at 1, 2, and 4 weeks after allo-SCT. (C) Frequency of YFP expression within CD4+GM-CSF+ and CD8+GM-CSF+ T cells from colon and mLN at 1, 2, and 4 weeks after allo-SCT. (D) Total number of CD4+ GM-CSF+YFP+, GM-CSF+YFP−, and total CD4+ T cells in colon and mLN at 1, 2, and 4 weeks after allo-SCT. (E) Total number of CD8+ GM-CSF+YFP+, GM-CSF+YFP−, and total CD8+ T cells in colon and mLN at 1, 2, and 4 weeks after allo-SCT. (A-E) The data are combined from 3 replicate experiments (n = 14-16 mice per group). (F) Representative histograms showing BATF expression in RORγt+ and RORγt− CD4+ and CD8+ T cell fractions in colon 2 and 4 weeks after allo-SCT. (G) Representative histograms showing BATF expression in GM-CSF+ and GM-CSF− colonic CD4+ T cells fractions 2 and 4 weeks after allo-SCT. (H) Batf relative gene expression in YFP+ and YFP− colonic CD4+ T cells 4 weeks after allo-SCT (n = 6 per group). Statistical analyses were performed by unpaired Student t test when comparing between 2 groups and 1-way analysis of variance when comparing over time (mean ± standard error of the mean [SEM]). *P < .05; **P < .01; ***P < .001; ****P < .0001.
Th17/Tc17 are a significant source of GM-CSF in the GI tract after allo-SCT. Cytokine and transcription factor expression by CD4+YFP+ and CD8+YFP+ T cells was assessed in the colon and mLN 1, 2, and 4 weeks after allogeneic (B6.IL-17CreRosa26eYFP→Balb/c) transplant. (A) Representative flow cytometry plots gated on isotype controls demonstrating GM-CSF expression in YFP+ vs YFP− T cells 4 weeks after allo-SCT. (B) Frequency of GM-CSF expression in CD4+ and CD8+ T cells within YFP+ and YFP− compartments from colon and mLN at 1, 2, and 4 weeks after allo-SCT. (C) Frequency of YFP expression within CD4+GM-CSF+ and CD8+GM-CSF+ T cells from colon and mLN at 1, 2, and 4 weeks after allo-SCT. (D) Total number of CD4+ GM-CSF+YFP+, GM-CSF+YFP−, and total CD4+ T cells in colon and mLN at 1, 2, and 4 weeks after allo-SCT. (E) Total number of CD8+ GM-CSF+YFP+, GM-CSF+YFP−, and total CD8+ T cells in colon and mLN at 1, 2, and 4 weeks after allo-SCT. (A-E) The data are combined from 3 replicate experiments (n = 14-16 mice per group). (F) Representative histograms showing BATF expression in RORγt+ and RORγt− CD4+ and CD8+ T cell fractions in colon 2 and 4 weeks after allo-SCT. (G) Representative histograms showing BATF expression in GM-CSF+ and GM-CSF− colonic CD4+ T cells fractions 2 and 4 weeks after allo-SCT. (H) Batf relative gene expression in YFP+ and YFP− colonic CD4+ T cells 4 weeks after allo-SCT (n = 6 per group). Statistical analyses were performed by unpaired Student t test when comparing between 2 groups and 1-way analysis of variance when comparing over time (mean ± standard error of the mean [SEM]). *P < .05; **P < .01; ***P < .001; ****P < .0001.
GM-CSF receptor signaling promotes alloantigen presentation by donor DC
Given that GM-CSF is important to cells reconstituting from the donor stem cell compartment that are involved in antigen presentation (eg, DCs, macrophages), we hypothesized that the pathogenic pathway invoked by this cytokine may be through enhancement of donor-derived antigen-presenting cell (APC) function.8,16 To undertake these analyses, we used our well-established T-cell receptor transgenic models to report indirect alloantigen presentation by migratory, CCR7-dependent, colon-derived DCs, which unlike donor macrophages, we have found to be critical for fulminant gut GVHD.15,22 In these systems, luciferase-expressing TEa T-cell receptor-transgenic T cells and the YAe antibody reports for alloantigen (MHC Class II I-E) presented by donor dendritic cells10 (an experimental system extensively detailed in Koyama et al15 ). We undertook B6 (CD45.1+I-Ab) → BALB/c (CD45.2+I-Ad/I-Ed) transplants with wild-type (WT) or common β chain-deficient (βc−/−) donor grafts and quantified DC numbers after reconstitution at day 15. The numbers of DC in the mLN and peripheral lymph node were significantly reduced in the absence of common β chain signaling (Figure 2A; supplemental Figure 1A). The DC in the mLN also had reduced levels of CCR7 expression (Figure 2B) and were presenting lower amounts of alloantigen, quantified by YAe antibody (Figure 2C). Thus, there both were reduced numbers of donor DC and they were presenting less alloantigen on a per cell basis. To understand how alloantigen presentation by donor DC was affected by GM-CSF signaling in the GI tract, we further quantified alloantigen presentation by known DC subsets in the mLN. As previously described,15 high numbers of (YAe+) alloantigen-presenting APCs were seen in the CD103+ DC subsets, and alloantigen-presenting DCs were profoundly reduced in all fractions in the absence of GM-CSF signaling through the common β chain (Figure 2D). Interestingly, alloantigen presentation on a per cell basis (quantification of YAe expression) was reduced in the CD103+CD11b− and the CD103−CD11b+ DC fraction in mLN, independent of any reductions in major histocompatibility complex (MHC) class II expression (supplemental Figure 1B). Next, we quantified the biological relevance of these phenotypic changes by infusing alloantigen-specific TEa T cells and tracking their expansion 3 days later by luciferase imaging.15,17 As predicted, in the absence of common β chain signaling in bone marrow-derived DC, alloantigen-specific T-cell priming and expansion was significantly impaired in the mLN (Figure 2E). Next, we transplanted BALB/c mice with donor TEa T cells that could only respond to alloantigen presentation by donor DC15,17 where GM-CSF signaling through the common β chain was either intact or absent. In the latter setting, the attenuation in donor DC expansion and alloantigen presentation (since Tea T cells cannot respond to recipient MHC class II) was associated with a significant reduction in acute GVHD histopathology within the colon (Figure 2F).
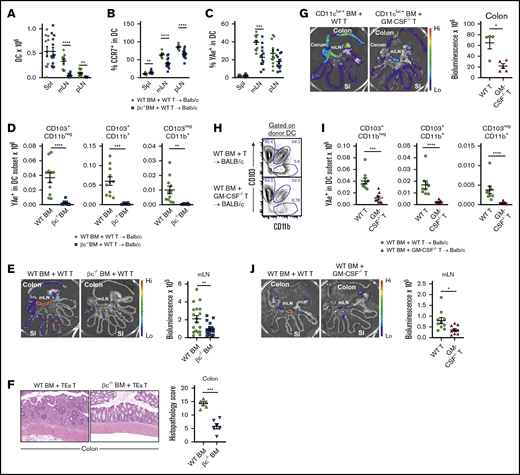
Common β-chain signaling drives alloantigen presentation in the GI tract. (A-E) BALB/c mice were transplanted with TCD BM from B6.WT or B6.common β-chain−/− mice and B6.WT T cells. On day 12 B6.TEaluc+ cells were injected, and 3 days later, DC subsets and TEa T-cell expansion were analyzed. The absolute numbers of total donor DC (A), the frequency of CCR7+ (B), and the frequency of YAe+ in total donor DC (C) from spleen, mLN, and peripheral lymph node are shown. (D) The absolute number of YAe+ donor DC within mLN. (A-D) The data are combined from 2 replicate experiments (n = 11-13 per group). (E) Representative images (left) and quantification (right) of bioluminescence signals as a measure of alloantigen presentation and T-cell priming. The data are combined from 3 replicate experiments (n = 18-20 per group). (F) BALB/c mice were transplanted with B6.WT or B6.common β-chain−/− TCD BM and 2 × 106 sort purified CD4+ B6.TEa T cells. Representative hematoxylin and eosin images (left) and semiquantitative histopathology scores of colon (right) at day 14 after BMT (n = 5-6 per group). (G) BALB/c mice were transplanted with TCD BM from B6.CD11cluc+ reporter mice and B6.WT or B6.GM-CSF−/− T cells. On day 18 donor DC (CD11cluc+) were quantified (n = 4-5 per group). (H-J) BALB/c mice were transplanted with TCD BM from B6.WT mice and B6.WT or B6.GM-CSF−/− T cells. On day 12 TEaluc+ cells were injected, and 3 days later, DC subsets and TEa expansion were analyzed. Representative fluorescence-activated cell sorter plots of DC subset proportions (H), absolute numbers of YAe+ donor DC subsets in mLN (I), and representative images (left) and quantification (right) of bioluminescence signals as a measure of alloantigen presentation and T-cell priming (J). The data are combined from 2 replicate experiments (n = 8-9 per group). Statistical analysis by unpaired Student t test with Welch’s correction (mean ± SEM). *P < .05; **P < .01; ***P < .001; ****P < .0001.
Common β-chain signaling drives alloantigen presentation in the GI tract. (A-E) BALB/c mice were transplanted with TCD BM from B6.WT or B6.common β-chain−/− mice and B6.WT T cells. On day 12 B6.TEaluc+ cells were injected, and 3 days later, DC subsets and TEa T-cell expansion were analyzed. The absolute numbers of total donor DC (A), the frequency of CCR7+ (B), and the frequency of YAe+ in total donor DC (C) from spleen, mLN, and peripheral lymph node are shown. (D) The absolute number of YAe+ donor DC within mLN. (A-D) The data are combined from 2 replicate experiments (n = 11-13 per group). (E) Representative images (left) and quantification (right) of bioluminescence signals as a measure of alloantigen presentation and T-cell priming. The data are combined from 3 replicate experiments (n = 18-20 per group). (F) BALB/c mice were transplanted with B6.WT or B6.common β-chain−/− TCD BM and 2 × 106 sort purified CD4+ B6.TEa T cells. Representative hematoxylin and eosin images (left) and semiquantitative histopathology scores of colon (right) at day 14 after BMT (n = 5-6 per group). (G) BALB/c mice were transplanted with TCD BM from B6.CD11cluc+ reporter mice and B6.WT or B6.GM-CSF−/− T cells. On day 18 donor DC (CD11cluc+) were quantified (n = 4-5 per group). (H-J) BALB/c mice were transplanted with TCD BM from B6.WT mice and B6.WT or B6.GM-CSF−/− T cells. On day 12 TEaluc+ cells were injected, and 3 days later, DC subsets and TEa expansion were analyzed. Representative fluorescence-activated cell sorter plots of DC subset proportions (H), absolute numbers of YAe+ donor DC subsets in mLN (I), and representative images (left) and quantification (right) of bioluminescence signals as a measure of alloantigen presentation and T-cell priming (J). The data are combined from 2 replicate experiments (n = 8-9 per group). Statistical analysis by unpaired Student t test with Welch’s correction (mean ± SEM). *P < .05; **P < .01; ***P < .001; ****P < .0001.
Because IL-3 and IL-5 also signal through the common β chain receptor, we confirmed these defects in alloantigen-presenting DC were specifically the result of donor T-cell–derived GM-CSF secretion by repeating these transplants with T cells that were GM-CSF deficient (GM-CSF−/−). We have shown that donor antigen presentation in the mLN is mediated by donor DCs that expand initially in the colon and then migrate to the mLN via CCR7.15 Here, we used B6.CD11c-luciferase reporter bone marrow to determine colon donor DC expansion in the presence of B6 WT vs GM-CSF−/− T cells. We demonstrate that in the absence of GM-CSF secretion by donor T cells, the pathogenic expansion of donor DCs in the colon is averted (Figure 2G). In the presence of GM-CSF−/− T cells, the greatest proportional reductions were noted in the CD103+CD11b+ DC subsets (Figure 2H; supplemental Figure 2A), but all alloantigen-presenting donor DC subsets were profoundly reduced in the mLN (Figure 2I). Alloantigen presentation on a per cell basis was reduced in CD103+CD11b+ and the CD103−CD11b+ DC fractions in mLN, independent of any reductions in MHC class II expression (supplemental Figure 2B). Finally, we confirmed that the absence of GM-CSF secretion from the donor T cell resulted in reduced antigen presentation and donor T-cell priming in the mLN (Figure 2J), phenocopying the findings seen when bone marrow-derived DC lacked the ability to signal GM-CSF through the common β chain receptor.
Discussion
We show that GM-CSF is secreted by donor T cells of both Th17 and non-Th17 fates, and that these cells preferentially accumulate in the colon after BMT. Within the GI tract, we define a novel mechanism by which donor T cells promote donor APC expansion and antigen presentation via donor DCs, which in turn amplify donor T-cell function within the mLN in a feed-forward cascade to drive lethal GVHD.
Although GM-CSF–producing donor T cells have now been widely reported as pathogenic,5,-7 their origin and the molecular factors controlling developmental specificity have been less clear. We noted that a significant minority of GM-CSF-secreting T cells were of the Th17 lineage which are characterized by high RORγt expression.23 One recent study demonstrated that GM-CSF+CD4+ T cells do not develop after BMT in the absence of the transcription factor BATF,7 which is perhaps not surprising, given the critical role of this transcription factor in T-cell activation24 and T cell (particularly Th17 and Tfh) differentiation.25 Consistent with this, we noted universally high BATF expression in all donor T cells after allo-SCT. Furthermore, as CD8+ T-cell proliferation and function is reported to be highly BATF dependent,26 donor BATF-deficient grafts may also result in impaired CD8+ T-cell responses and contribute to significant changes in transplant outcome. It is thus likely that additional factors are involved, particularly in the RORγtneg Th17-independent GM-CSF secreting donor T-cell lineage after SCT, and this would seem an important focus for further studies.
There are likely to be multiple mechanisms by which GM-CSF-secreting donor T cells promote acute GVHD in the GI tract, which include direct effector function (ie, cytokine)6,7 together with their described ability to generate proinflammatory myeloid populations.5 Here we used well-established experimental systems that precisely allow the quantification of alloantigen presentation by donor DCs.15,17 These systems have definitively demonstrated that during GVHD, donor CD103+ donor DC accumulate in the colon and migrate to the mLN under the guidance of CCR7 and prime donor CD4 T cells, while imprinting them with integrins that guide them to the GI tract to mediate lethal GVHD.15,16 Using these same systems, we intriguingly demonstrate that the DC component of this pathway can be initiated by GM-CSF derived from the donor T cell. Thus, GM-CSF from the donor T cell is required for maximal donor DC expansion and antigen presentation in the GI tract. The differences in alloantigen presentation on a per cell basis seen in DC subsets in recipients of βc−/− BM vs GM-CSF−/− donor T cells likely reflects the fact that there are additional sources of GM-CSF beyond donor T cells, whereas other common-β chain signaling cytokines (eg, IL-3/5) may also play a role.
One note of caution in regard to GM-CSF inhibition after BMT is that it has also been shown to be important in maintaining regulatory T-cell homeostasis and inhibiting chronic GVHD late after SCT.27 This effect is presumably via the maintenance of the MHC class II antigen presentation in the periphery that is known to be critical for regulatory T-cell survival.28 Thus, these data provide a logical rationale to administer GM-CSF blockade early after BMT to prevent acute GVHD, either prophylactically in conjunction with standard immune suppression or in patients identified high-risk by current biomarker-based approaches.29
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank staff in the Flow Cytometry Facility at QIMR-Berghofer for cell sorting and Madeleine Flynn for graphic design.
This work was supported by funding from the National Health and Medical Research Council (G.R.H.).
Authorship
Contribution: K.H.G. and M.K. designed and performed experiments, analyzed data, and helped write the manuscript; K.E.L., K.C., K.S.E., R.D.K., A.S.H., and L.D.S. performed experiments and/or analyzed data; A.D.C. provided blinded histological assessment of GVHD pathology; A.F.L. provided critical reagents and reviewed the manuscript; K.P.A.M. provided intellectual input and reviewed the manuscript; and G.R.H. provided intellectual input and helped write the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Kate H. Gartlan, QIMR Berghofer Medical Research Institute, 300 Herston Rd, Herston, QLD 4006, Australia; e-mail: kate.gartlan@qimrberghofer.edu.au.

