

Key Points
VEGFR1/2 kinase inhibitor and neutralizing anti-VEGFR2 antibody block PHD inhibitor FG-4497–mediated increase in HSPC mobilization.
The inhibitory effect of anti-VEGFR2 on HSPC mobilization is likely HSPC extrinsic, as VEGFR2 is only detected in BM endothelial cells.

Abstract
In normoxia, hypoxia-inducible transcription factors (HIFs) are rapidly degraded within the cytoplasm as a consequence of their prolyl hydroxylation by oxygen-dependent prolyl hydroxylase domain (PHD) enzymes. We have previously shown that hematopoietic stem and progenitor cells (HSPCs) require HIF-1 for effective mobilization in response to granulocyte colony-stimulating factor (G-CSF) and CXCR4 antagonist AMD3100/plerixafor. Conversely, HIF PHD inhibitors that stabilize HIF-1 protein in vivo enhance HSPC mobilization in response to G-CSF or AMD3100 in a cell-intrinsic manner. We now show that extrinsic mechanisms involving vascular endothelial growth factor receptor-2 (VEGFR2), via bone marrow (BM) endothelial cells, are also at play. PTK787/vatalanib, a tyrosine kinase inhibitor selective for VEGFR1 and VEGFR2, and neutralizing anti-VEGFR2 monoclonal antibody DC101 blocked enhancement of HSPC mobilization by FG-4497. VEGFR2 was absent on mesenchymal and hematopoietic cells and was detected only in Sca1+ endothelial cells in the BM. We propose that HIF PHD inhibitor FG-4497 enhances HSPC mobilization by stabilizing HIF-1α in HSPCs as previously demonstrated, as well as by activating VEGFR2 signaling in BM endothelial cells, which facilitates HSPC egress from the BM into the circulation.
Introduction
Hematopoietic stem and progenitor cell (HSPC) mobilization from the bone marrow (BM) into the blood is the mainstream procedure to harvest HSPCs for transplantation. Daily injection of granulocyte colony-stimulating factor (G-CSF) is the standard to elicit therapeutic HSPC mobilization in humans.1 The mechanisms of HSPC mobilization in response to G-CSF are complex. They involve indirect mechanisms in which the BM microenvironment and HSPC niches are altered, reducing HSPC retention within their BM niches together with some direct mechanisms promoting direct emigration of HSPCs out of their niches toward the circulation.2-8 We have recently demonstrated that 1 of these direct mechanisms involves the stabilization and activation of hypoxia-inducible transcription factor (HIF)-1.9 Indeed, conditional deletion of the Hif1a gene in mouse HSPCs abrogates their mobilization in response to G-CSF or AMD3100.9 In addition to HIF-1’s role in HSPC mobilization, conditional deletion of the Hif1a gene in hematopoietic and stromal compartments impairs hematopoietic stem cell (HSC) quiescence and self-renewal,10 whereas selective Hif1a deletion in hematopoietic cells does not impair HSC function.11 Genetic stabilization10 or pharmacological stabilization12 of HIFs increases HSC quiescence and reconstitution potential in vivo. HIF-1α protein abundance is posttranslationally regulated, in part, by oxygen in the extracellular milieu. In the presence of an O2 concentration > 5%, HIF-1α protein is rapidly degraded in the cytosol before its nuclear translocation.13 HIF-1α O2-dependent degradation is triggered by 3 HIF O2-dependent 4-prolyl hydroxylase domain (PHD) enzymes (HIF PHD 1-3) that hydroxylate specific proline residues within HIF-1α oxygen-dependent degradation domains.14-16 These 3 HIF PHD enzymes are Fe2+-dependent dioxygenases using α-ketoglutarate and oxygen as substrates. They can be inhibited in vitro and in vivo with selective small synthetic inhibitors, such as FG-4497, a modified isoquinoline linked to a carbonyl amino acetic acid17 that mimics and competes with α-ketoglutarate in HIF PHD catalytic center.18,19 FG-4497 selectively inhibits HIF PHD 1-3 enzymes with a 50% inhibitory concentration (IC50) between 0.2 and 0.3 µM,20 thereby preventing HIF-1α and HIF-2α prolylhydroxylation and subsequent ubiquitination and degradation by the von Hippel-Lindau complex. Stabilized HIF-1α and HIF-2α proteins complex to aryl hydrocarbon receptor nuclear translocator in the cytosol for subsequent nuclear translocation where HIFs can activate transcription of target genes.17 FG-4497 has a >100 to 200–fold higher IC50 (40 µM) for closely related HIF transmembrane prolyl 4-hydroxylase P4H-TM,20 but its activity against other α-ketoglutarate dioxygenases has not been reported.
We have previously demonstrated that FG-4497 and other HIF PHD inhibitors synergistically enhance HSPC mobilization in response to G-CSF or AMD31009 in the C57BL/6 inbred mouse strain, which mobilizes poorly in response to G-CSF21 and, therefore, may represent a model of poor mobilization. The lack of an FG-4497–mobilizing effect in mice with conditional deletion of the Hif1a gene in HSPCs confirmed that the promobilizing effect of FG-4497 was not an off-target effect; instead, it was mediated by HIF-1α, in part via an HSPC-intrinsic mechanism.9 Furthermore, in nonobese diabetic severe combined immune-deficient Il2rg−/− mice that were irradiated and transplanted with human cord blood CD34+ HSPCs, administration of FG-4497 partially rescued the mobilization defect in human HSPCs in response to G-CSF that is observed in these humanized mice.22 Although not completely understood, the promobilizing effect of HIF PHD inhibitors, such as FG-4497, may be mediated by reducing the CXCR4 intracellular pool in HSPCs and decreasing their chemotactic response to CXCL12.9 In addition to these HSPC-intrinsic mechanisms, there is evidence that HIF PHD inhibitors enhance HSPC mobilization via niche-mediated extrinsic mechanisms, because PHD inhibitors also reduce Cxcl12 messenger RNA (mRNA) expression by BM stromal cells in response to G-CSF.9
Considering that HIF-1 and HIF-2 are well known to activate the transcription of vascular endothelial growth factor-A (VEGF-A),23,24 Vegfa mRNA expression is increased in the endosteal region of the BM of mice treated with G-CSF,25 and chronic VEGF-A administration elicits HSPC mobilization in mice,26 we tested the hypothesis that the promobilizing effect of HIF PHD inhibitor FG-4497 on HSPCs in response to G-CSF involves VEGF-A and VEGF receptors (VEGFRs).
Materials and methods
Mice
All experiments were performed on 8- to 9-week-old C57BL/6 male mice purchased from the Animal Resource Centre (Perth, Australia) and approved by the University of Queensland Animal Ethics Committee.
In vivo treatments
VEGFR1 and VEGFR2 tyrosine kinase activity was blocked in vivo by gavaging mice with synthetic tyrosine kinase inhibitor PTK787/vatalanib27 (Biorbyt, Cambridge, United Kingdom; 20 mg/mL in polyethylene glycol 2000 [Fluka] at 37°C). Mice were gavaged daily with 100 µL/20 g body weight of this suspension, corresponding to 100 mg/kg vatalanib. Control mice were gavaged with an equivalent volume of vehicle.
HIF-1α protein was stabilized in vivo by injecting FG-4497 daily (20 mg/kg, intraperitoneally).9,12 Control animals were injected with an equivalent volume of 5% dextrose (weight to volume ratio) in water.
VEGFR2 was specifically inhibited in vivo using the neutralizing rat anti-mouse VEGFR2 monoclonal antibody (mAb) DC101.26 Purified endotoxin-free DC101 mAb (Bio-X-Cell, Lebanon, NH) was injected intraperitoneally (800 µg, every other day).26 Control mice were injected with 800 µg of isotype-matched rat IgG1 mAb clone HRPN (Bio-X-Cell).
Recombinant human G-CSF (filgrastim; Amgen, Thousand Oaks, CA) was injected twice daily (125 µg/kg, subcutaneously). Control mice were injected with saline.
Additional materials and methods used in this study are described in supplemental Methods.
Results
VEGFR kinase inhibitor PTK787 abrogates the promobilizing effect of FG-4497
To test the possible involvement of VEGF-A and VEGFRs in the promobilizing effect of the PHD inhibitor FG-4497 on HSPC mobilization in response to G-CSF, C57BL/6 mice were mobilized with FG-4497 or vehicle for 3 days, together with G-CSF for the last 2 days (Figure 1A); we have previously shown that this schedule further enhances HSPC mobilization in response to G-CSF.9 Half of the mice were also gavaged for 3 consecutive days with vehicle or PTK787 (vatalanib), which selectively inhibits VEGFR1 and VEGFR2 tyrosine kinase activities (IC50 = 77 and 37 pM, respectively) and, to a lesser degree, VEGFR3, PDGFR-β, and c-Kit tyrosine kinase activities (IC50 = 660, 730, and 580 pM, respectively27 ).
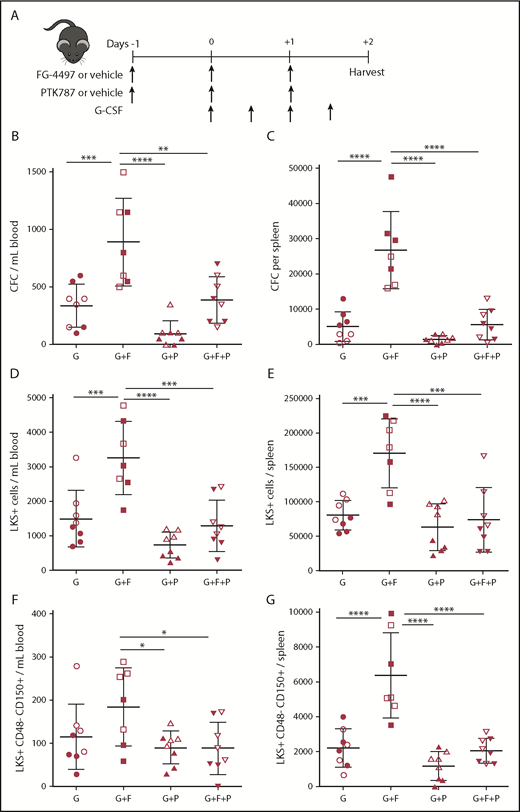
VEGFR tyrosine kinase inhibitor PTK787 inhibits synergistic effect of FG-4497 on G-CSF–induced HSPC mobilization. (A) Mice were administered G-CSF for 2 days, FG-4497 or vehicle for 3 days, and PTK787 or vehicle for 3 days. At harvest, the numbers of CFCs (B-C), LKS+ HSPCs (D-E), and LKS+CD48−CD150+ HSCs (F-G) were measured in peripheral blood (B,D,F) and spleen (C,E,G). Data are from 2 pooled experiments performed several months apart. Each point represents an individual mouse. Bars are means ± standard deviation. *P < .05, **P < 10−2, ***P < 10−3, ****P < 10−4. G, G-CSF with vehicles; G+F, G-CSF plus FG-4497; G+F+P, G-CSF plus FG-4497 plus PTK787; G+P, G-CSF plus PTK787.
VEGFR tyrosine kinase inhibitor PTK787 inhibits synergistic effect of FG-4497 on G-CSF–induced HSPC mobilization. (A) Mice were administered G-CSF for 2 days, FG-4497 or vehicle for 3 days, and PTK787 or vehicle for 3 days. At harvest, the numbers of CFCs (B-C), LKS+ HSPCs (D-E), and LKS+CD48−CD150+ HSCs (F-G) were measured in peripheral blood (B,D,F) and spleen (C,E,G). Data are from 2 pooled experiments performed several months apart. Each point represents an individual mouse. Bars are means ± standard deviation. *P < .05, **P < 10−2, ***P < 10−3, ****P < 10−4. G, G-CSF with vehicles; G+F, G-CSF plus FG-4497; G+F+P, G-CSF plus FG-4497 plus PTK787; G+P, G-CSF plus PTK787.
PTK787 administration, together with G-CSF and FG-4497, abrogated the enhancing effect of FG-4497 on G-CSF–induced mobilization of colony-forming cells (CFCs), Lin−Kit+Sca1+ (LKS+) immature HSPCs, and phenotypic LKS+CD48−CD150+ HSCs, despite the fact that PTK787 did not alter HSPC mobilization in response to G-CSF alone (Figure 1B-G; supplemental Figure 1). This suggests that tyrosine kinases inhibited by PTK787 are involved in increased HSPC mobilization in response to FG-4497.
VEGF-A increases in G-CSF–mobilized BM, and VEGFR1 and VEGFR2 are expressed by endothelial cells in the BM
PTK787 is a potent inhibitor of VEGFR1 and VEGFR2 tyrosine kinases, and both of these kinases are activated by the binding of their common ligand VEGF-A.28,29 We found that VEGF-A protein concentration was increased in mouse BM fluids culminating at day 4 of G-CSF administration (Figure 2A). To determine whether VEGF-A receptors are expressed on HSPCs, we sorted LKS+ cells from the BM of mice treated for 2 days with saline or G-CSF, extracted RNA, and performed RNA-sequencing (RNA-seq). Although mRNA for Kit, Ly6a (encoding Sca1 antigen), and Flt3 was abundantly expressed as expected, mRNA for Flt1 (VEGFR1), Kdr (VEGFR2), Flt4 (VEGFR3), and Vegfa was not detected in LKS+ cells in steady-state or mobilized BM (Figure 2B). Our RNA-seq results are in agreement with expression microarray data from 39 sorted mouse HSPC subsets in the Gene Expression Commons database30 (supplemental Figure 2). Flt1, Kdr, and Flt4 mRNA was exclusively expressed by CD45−Ter119−CD51−Tie2+ endothelial cells, whereas Vegfa was absent from all cell types in this database (data not shown). To further refine this analysis at the single-cell level, we used the Single-Cell Gene Expression Atlas (http://blood.stemcells.cam.ac.uk/single_cell_atlas.html), which quantifies mRNA in single cells in the full spectrum of HSPCs, from HSCs to myeloid progenitor cells.31 The distributions of Lin−Kit+Sca1− (LKS−) hematopoietic progenitor cells (HPCs), LKS+ HSPCs, LKS+CD34−Flt3− HSCs, and Slamf1 (CD150) and Procr (endothelial protein C receptor CD201) mRNA, which is expressed by HSCs,32,33 are shown to illustrate the different HSPC subsets. Flt1 and Flt4 mRNA was detected in very rare single cells, whereas Kdr mRNA was detected in few cells within the HSC region (Figure 2C). Vegfa mRNA was detected in few cells scattered through the entire HPC spectrum.
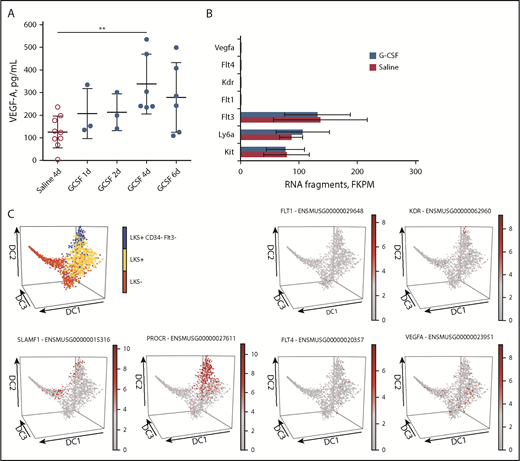
VEGF-A protein increased in BM fluids during G-CSF treatment, but VEGFR1 (Flt1), VEGFR2 (Kdr), and VEGFR3 (Flt4) mRNA are not detected in HSPCs. (A) VEGF-A concentration in BM fluids from mice mobilized with G-CSF. Data are mean ± standard deviation of 3 to 9 mice per time point. (B) RNA-seq analysis of LKS+ cells sorted from the BM of mice treated with saline or G-CSF for 2 days. Results in fragments per kilobase of transcript per million mapped reads (FKPM) are from 3 mice for each treatment group (mean ± standard deviation). (C) In silico single-cell clustering of expression of Flt1, Kdr, Flt4, and Vegfa mRNA on Single-Cell Gene Expression Atlas Web site (http://blood.stemcells.cam.ac.uk/single_cell_atlas.html). Distribution of individual HSPC subsets in blue (HSCs), yellow (LKS+ HSPCs), and red (LKS− HPCs) (upper left panel). In all other panels, the intensity of the mRNA signal increases from gray (negative) to red. Slamf1 (CD150) and Procr (EPCR) mRNA identify long-term reconstituting HSCs. Among all single HSPCs sequenced (gray dots), the red dots represent single cells positive for the indicated mRNA. **P < .01.
VEGF-A protein increased in BM fluids during G-CSF treatment, but VEGFR1 (Flt1), VEGFR2 (Kdr), and VEGFR3 (Flt4) mRNA are not detected in HSPCs. (A) VEGF-A concentration in BM fluids from mice mobilized with G-CSF. Data are mean ± standard deviation of 3 to 9 mice per time point. (B) RNA-seq analysis of LKS+ cells sorted from the BM of mice treated with saline or G-CSF for 2 days. Results in fragments per kilobase of transcript per million mapped reads (FKPM) are from 3 mice for each treatment group (mean ± standard deviation). (C) In silico single-cell clustering of expression of Flt1, Kdr, Flt4, and Vegfa mRNA on Single-Cell Gene Expression Atlas Web site (http://blood.stemcells.cam.ac.uk/single_cell_atlas.html). Distribution of individual HSPC subsets in blue (HSCs), yellow (LKS+ HSPCs), and red (LKS− HPCs) (upper left panel). In all other panels, the intensity of the mRNA signal increases from gray (negative) to red. Slamf1 (CD150) and Procr (EPCR) mRNA identify long-term reconstituting HSCs. Among all single HSPCs sequenced (gray dots), the red dots represent single cells positive for the indicated mRNA. **P < .01.
We next performed flow cytometry with direct fluorescent conjugates of rat anti-mouse VEGFR1 and anti-mouse VEGFR2 mAbs to detect them at the surface of adult mouse BM HSPCs. In pilot experiments, we tested these antibodies on the immortalized mouse yolk sac endothelial cell line C16634 and the mouse mesenchymal progenitor cell line Kusa4b10.35 As expected, anti-VEGFR1 and anti-VEGFR2 antibodies bound to C166 endothelial cells but not to Kusa4b10 mesenchymal cells (supplemental Figure 3). When used on BM leukocytes, neither VEGFR1 nor VEGFR2 proteins were detected at the surface of any HSPC subset, including HSCs, by flow cytometry (supplemental Figure 4).
Because some macrophage subsets reportedly express endothelial receptor tyrosine kinase receptors, such as Tie2,36 and granulocytes2,3 and BM-resident macrophages37,38 regulate the levels of CXCL12 and VCAM-1 protein and mRNA expression in BM hematopoietic niches in the BM, whereas subsets of blood granulocytes express VEGFR1 but not VEGFR2,39 we also assessed VEGFR1 and VEGFR2 proteins on BM granulocytes and macrophage subsets that we could identify using a combination of anti-F4/80, anti-Ly6G, CD169, anti–VCAM-1, and CD11b antibodies (supplemental Figure 4). With the exception of the CD11b+F4/80+Ly6G+VCAM1+CD169+ population, which had 0.2% cells positive for VEGFR1 and VEGFR2, VEGFR1 and VEGFR2 were not detected on the other BM myeloid subsets (supplemental Figure 5), consistent with data from the Gene Expression Commons database (supplemental Figure 2).
To measure VEGFR1 and VEGFR2 expression on BM stromal cells, we first enriched BM endosteal cells in nonhematopoietic stromal cells by magnetic depletion of CD45+ leukocytes and Ter119+ erythroid cells and then stained CD45−Ter119−CD31+ endothelial cells and CD45−Ter119−CD31− mesenchymal cells with VEGFR1 and VEGFR2 mAbs. VEGFR2 was primarily expressed on Sca1+ endothelial cells (CD45−Ter119−CD31+Sca1+) (Figure 3). VEGFR2 was not expressed on mesenchymal cells, such as CD45−Ter119−CD31−PDGFRα+Sca1+ cells, CD45−Ter119−CD31−PDGFRα+Sca1− cells (also defined as CXCL12-abundant reticular cells40 ), or CD45−Ter119−CD31−PDGFRα−Sca1− cells (Figure 3). VEGFR1 was detected on the subset of Sca1+ endothelial cells also expressing VEGFR2, as well as on a small percentage of CD45−Ter119−CD31−PDGFRα+Sca1− cells. Our flow cytometry results for VEGFR2 are consistent with the expression of Kdr mRNA exclusively in CD45−Tie2+ endothelial cells in the Gene Expression Commons database (supplemental Figure 2); further validation by real-time quantitative reversed transcription polymerase chain reaction (qRT-PCR) would be needed to confirm the flow cytometry detection of VEGFR1 on mesenchymal cells.
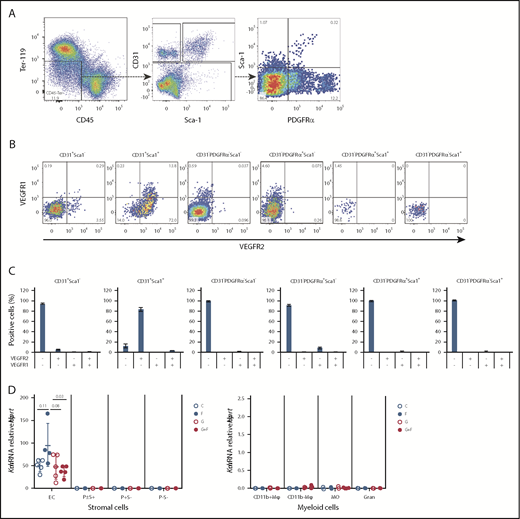
VEGFR1 and VEGFR2 are expressed by Sca1+endothelial cells in the BM. BM cells were extracted from the endosteal region, enriched in stromal cells by magnetic depletion of CD45+ leukocytes and Ter119+ erythroid cells, and stained for hematopoietic, endothelial, and mesenchymal markers together with anti-VEGFR1 and anti-VEGFR2 fluorescent antibodies. (A) Gating strategy to identify endothelial and mesenchymal cells. Following the exclusion of residual CD45+ and Ter119+ leukocytes and erythroid cells, nonhematopoietic CD45−Ter119− cells were gated as CD31+Sca1− endothelial cells and CD31+Sca1+ endothelial cells. Remaining CD45−Ter119−CD31− mesenchymal cells were gated as PDGFRα−Sca1−, PDGFRα+Sca1−, PDGFRα+Sca1+, and PDGFRα−Sca1+ cells. (B) Typical dot-plots showing VEGFR1 and VEGFR2 expression on previously defined endothelial and mesenchymal populations. (C) Percentage of endothelial and mesenchymal BM cells expressing VEGFR1 and/or VEGFR2. Bar graphs show mean ± standard deviation of 4 mice. (D) Kdr (VEGFR2) mRNA expression by qRT-PCR on CD11b+F4/80+VCAM1+CD169+ macrophages (CD11b+Mφ), CD11b−F4/80+VCAM1+CD169+ macrophages (CD11b-Mφ), CD11b+F4/80+VCAM1−CD169− monocytes (MO), CD11b+F4/80−Ly6G+ granulocytes (Gran), CD45−Ter119−CD31+ endothelial cells (EC), CD45−Ter119−CD31−PDGFRα+/−Sca1+ mesenchymal progenitor cells (P±S+), PDGFRα+Sca1− mesenchymal progenitor cells (P+S-), and CD45−Ter119−CD31−PDGFRα−Sca1− stromal cells (P-S-) sorted from the BM of mice treated with saline (C), FG-4497 alone (F), G-CSF (G), or G-CSF plus FG-4497 (G+F) for 3 days. Each dot represents a separate mouse and separate sort. Data are relative to Hprt mRNA. The P values were calculated using ANOVA.
VEGFR1 and VEGFR2 are expressed by Sca1+endothelial cells in the BM. BM cells were extracted from the endosteal region, enriched in stromal cells by magnetic depletion of CD45+ leukocytes and Ter119+ erythroid cells, and stained for hematopoietic, endothelial, and mesenchymal markers together with anti-VEGFR1 and anti-VEGFR2 fluorescent antibodies. (A) Gating strategy to identify endothelial and mesenchymal cells. Following the exclusion of residual CD45+ and Ter119+ leukocytes and erythroid cells, nonhematopoietic CD45−Ter119− cells were gated as CD31+Sca1− endothelial cells and CD31+Sca1+ endothelial cells. Remaining CD45−Ter119−CD31− mesenchymal cells were gated as PDGFRα−Sca1−, PDGFRα+Sca1−, PDGFRα+Sca1+, and PDGFRα−Sca1+ cells. (B) Typical dot-plots showing VEGFR1 and VEGFR2 expression on previously defined endothelial and mesenchymal populations. (C) Percentage of endothelial and mesenchymal BM cells expressing VEGFR1 and/or VEGFR2. Bar graphs show mean ± standard deviation of 4 mice. (D) Kdr (VEGFR2) mRNA expression by qRT-PCR on CD11b+F4/80+VCAM1+CD169+ macrophages (CD11b+Mφ), CD11b−F4/80+VCAM1+CD169+ macrophages (CD11b-Mφ), CD11b+F4/80+VCAM1−CD169− monocytes (MO), CD11b+F4/80−Ly6G+ granulocytes (Gran), CD45−Ter119−CD31+ endothelial cells (EC), CD45−Ter119−CD31−PDGFRα+/−Sca1+ mesenchymal progenitor cells (P±S+), PDGFRα+Sca1− mesenchymal progenitor cells (P+S-), and CD45−Ter119−CD31−PDGFRα−Sca1− stromal cells (P-S-) sorted from the BM of mice treated with saline (C), FG-4497 alone (F), G-CSF (G), or G-CSF plus FG-4497 (G+F) for 3 days. Each dot represents a separate mouse and separate sort. Data are relative to Hprt mRNA. The P values were calculated using ANOVA.
To confirm our flow cytometry results, CD11b+ macrophages, CD11b− macrophages, monocytes, granulocytes, endothelial cells, and mesenchymal progenitor cells were sorted from the BM of mice treated with saline or FG-4497 alone for 3 days (n = 5 per group). The next day, a similar sorting was performed using cells from mice treated with G-CSF alone or in combination with FG-4497. Kdr mRNA was exclusively detected in CD45−Ter119−CD31+ endothelial cells and was absent from all myeloid and mesenchymal populations tested (Figure 3D). Furthermore, we observed that treatment with FG-4497 alone caused a trend toward higher Kdr expression in BM endothelial cells compared with vehicle-treated animals.
Therefore, in the adult mouse BM, VEGFR2 could only be detected in Sca1+ endothelial cells, and it was expressed at a much higher frequency than VEGFR1 on this subset.
In vivo neutralization of VEGFR2 inhibits the promobilizing effect of FG-4497 in response to G-CSF
Because VEGFR2 is the most abundantly expressed VEGFR in the BM, we neutralized VEGFR2 in vivo by administering the rat anti-mouse VEGFR2 mAb DC101 to mice mobilized with G-CSF alone or G-CSF plus FG-4497 (Figure 4A). Control mice were administered the isotype-matched rat IgG1 HRPN. Mice treated with the combination of G-CSF plus FG-4497 and control rat IgG1 mobilized CFCs, LKS+ HPCs, and phenotypic HSCs into blood and spleens significantly more than in other treatment groups. In particular, treatment with the combination of G-CSF plus FG-4497 and anti-VEGFR2 mAb DC101 returned mobilization of these cells to levels equivalent to the G-CSF–alone group (Figures 4B-G; supplemental Figure 6). Therefore, in vivo neutralization of VEGFR2 with blocking mAb DC101 inhibited most of the HSPC mobilization enhancement caused by FG-4497.
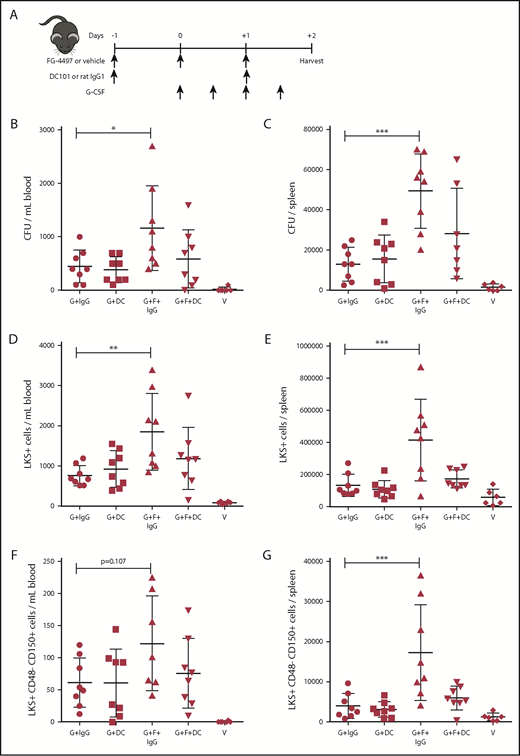
Neutralization of VEGFR2 blocks the effect of FG-4497 on G-CSF–induced HSPC mobilization. (A) Mice were administered G-CSF for 2 days, FG-4497 or vehicle for 3 days, and neutralizing rat anti-mouse VEGFR2 mAb DC101 or control rat IgG1 every other day. At harvest, the numbers of CFCs (B-C), LKS+ HSPCs (D-E), and LKS+CD48−CD150+ HSCs (F-G) were measured in peripheral blood (B,D,F) and spleen (C,E,G). Data are from 2 pooled experiments performed several months apart. Each point is an individual mouse. Bars are means ± standard deviation. *P < .05, **P < 10−2, ***P < 10−3. G+DC, G-CSF with vehicle DC101 mAb; G+F+DC, G-CSF plus FG-4497 plus DC101; G+F+IgG, G-CSF plus FG-4497 plus rat IgG1; G+IgG, G-CSF with vehicle with rat IgG1.
Neutralization of VEGFR2 blocks the effect of FG-4497 on G-CSF–induced HSPC mobilization. (A) Mice were administered G-CSF for 2 days, FG-4497 or vehicle for 3 days, and neutralizing rat anti-mouse VEGFR2 mAb DC101 or control rat IgG1 every other day. At harvest, the numbers of CFCs (B-C), LKS+ HSPCs (D-E), and LKS+CD48−CD150+ HSCs (F-G) were measured in peripheral blood (B,D,F) and spleen (C,E,G). Data are from 2 pooled experiments performed several months apart. Each point is an individual mouse. Bars are means ± standard deviation. *P < .05, **P < 10−2, ***P < 10−3. G+DC, G-CSF with vehicle DC101 mAb; G+F+DC, G-CSF plus FG-4497 plus DC101; G+F+IgG, G-CSF plus FG-4497 plus rat IgG1; G+IgG, G-CSF with vehicle with rat IgG1.
To further assess the effect of DC101 in vivo, we measured the concentration of VEGF-A, the main ligand of VEGFR2,29 in the blood plasma and BM fluids of these mice (Figure 5). Two-day G-CSF treatment increased VEGF-A concentration in the blood slightly (albeit nonsignificantly by 1-way analysis of variance [ANOVA]), irrespective of whether FG-4497 was present (Figure 5A). Remarkably, DC101 administration caused a fourfold increase in blood VEGF-A concentration. A similar pattern was observed in BM fluids (Figure 5B). Therefore, VEGF-A binding to VEGFR2 may contribute to a large proportion of VEGF-A internalization and elimination by endothelial cells, consistent with increased VEGFR2 expression in endothelial cells in response to FG-4497 (Figure 3D) or, alternatively, a compensation feedback due to VEGFR2 signaling blockade may take place, leading to enhanced VEGF-A production.

VEGF-A concentration is increased in vivo in response to DC101 and in vitro in response to FG-4497. (A-B) Mice were treated with G-CSF, FG-4497, neutralizing rat anti-mouse VEGFR2 mAb DC101, or control rat IgG1, as described in Figure 4A. Mouse VEGF-A protein concentration was measured by enzyme-linked immunosorbent assay in blood plasma (A) and femoral BM fluids (B). Data are from 2 pooled experiments performed at a 1-month interval. Each point represents an individual mouse. (C) FG-4497 stimulated VEGF-A secretion by BMDMs in vitro. BMDMs were cultured for 48 hours in the presence of 100 ng/mL rhuG-CSF (G), 40 µM FG-4497 (F), or both (G+F). Control cells were added vehicle. Supernatants were harvested at 24 and 48 hours, and mouse VEGF-A concentration was measured by enzyme-linked immunosorbent assay. Data are means ± SD of 3 replicates per conditions. (D) Western blot for HIF-1α on BMDMs cultured for 24 or 48 hours in the presence of 100 ng/mL rhuG-CSF or 40 µM FG-4497. (E) Vegfa, Vegfc, and Vegfd mRNA expression by qRT-PCR on CD11b+F4/80+VCAM1+CD169+ macrophages (CD11b+Mφ), CD11b−F4/80+VCAM1+CD169+ macrophages (CD11b-Mφ), CD11b+F4/80+VCAM1−CD169− monocytes (MO), CD11b+F4/80−Ly6G+ granulocytes (Gran), CD45−Ter119−CD31+ endothelial cells (EC), CD45−Ter119−CD31−PDGFRα+/−Sca1+ mesenchymal progenitor cells (P±S+), PDGFRα+Sca1− mesenchymal progenitor cells (P+S-), and CD45−Ter119−CD31−PDGFRα−Sca1− stromal cells (P-S-) sorted from the BM of mice treated with saline (C), FG-4497 alone (F), G-CSF (G), or G-CSF plus FG-4497 (G+F) for 3 days. Each circle represents a separate mouse and separate sort (n = 5 mice per population per treatment condition). Data are relative to Hprt mRNA. *P < .05, **P < 10−2, ***P < 10−3, ****P < 10−4, ANOVA.
VEGF-A concentration is increased in vivo in response to DC101 and in vitro in response to FG-4497. (A-B) Mice were treated with G-CSF, FG-4497, neutralizing rat anti-mouse VEGFR2 mAb DC101, or control rat IgG1, as described in Figure 4A. Mouse VEGF-A protein concentration was measured by enzyme-linked immunosorbent assay in blood plasma (A) and femoral BM fluids (B). Data are from 2 pooled experiments performed at a 1-month interval. Each point represents an individual mouse. (C) FG-4497 stimulated VEGF-A secretion by BMDMs in vitro. BMDMs were cultured for 48 hours in the presence of 100 ng/mL rhuG-CSF (G), 40 µM FG-4497 (F), or both (G+F). Control cells were added vehicle. Supernatants were harvested at 24 and 48 hours, and mouse VEGF-A concentration was measured by enzyme-linked immunosorbent assay. Data are means ± SD of 3 replicates per conditions. (D) Western blot for HIF-1α on BMDMs cultured for 24 or 48 hours in the presence of 100 ng/mL rhuG-CSF or 40 µM FG-4497. (E) Vegfa, Vegfc, and Vegfd mRNA expression by qRT-PCR on CD11b+F4/80+VCAM1+CD169+ macrophages (CD11b+Mφ), CD11b−F4/80+VCAM1+CD169+ macrophages (CD11b-Mφ), CD11b+F4/80+VCAM1−CD169− monocytes (MO), CD11b+F4/80−Ly6G+ granulocytes (Gran), CD45−Ter119−CD31+ endothelial cells (EC), CD45−Ter119−CD31−PDGFRα+/−Sca1+ mesenchymal progenitor cells (P±S+), PDGFRα+Sca1− mesenchymal progenitor cells (P+S-), and CD45−Ter119−CD31−PDGFRα−Sca1− stromal cells (P-S-) sorted from the BM of mice treated with saline (C), FG-4497 alone (F), G-CSF (G), or G-CSF plus FG-4497 (G+F) for 3 days. Each circle represents a separate mouse and separate sort (n = 5 mice per population per treatment condition). Data are relative to Hprt mRNA. *P < .05, **P < 10−2, ***P < 10−3, ****P < 10−4, ANOVA.
VEGF-A is produced by BM myeloid and stromal cells but has no direct chemotactic effect on HSPCs
Because VEGF-A is increased in the BM fluids of G-CSF–mobilized mice, particularly following DC101 mAb treatment, we sought to determine which cell type could be the source of enhanced VEGF-A protein in the BM. In silico analysis of the Gene Commons Database showed that Vegfa mRNA was not detected in sorted hematopoietic and stromal cells from the BM in the steady-state (data not shown), possibly due to low sensitivity of the probes used. Because we and other investigators have previously demonstrated that BM-resident macrophages regulate HSC niche function and mobilization,37,38,41 and in silico data from BioGPS revealed that bacterial lipopolysaccharides strongly stimulate Vegfa transcription in BM macrophages (supplemental Figure 7), mouse BM-derived macrophages (BMDMs) were cultured with mouse CSF-1 for 1 week, followed by 48 hours in the presence or absence of 100 ng/mL recombinant human G-CSF (rhuG-CSF) or 40 µM FG-4497. By enzyme-linked immunosorbent assay, FG-4497 strongly stimulated VEGF-A secretion by BMDMs in vitro, whereas G-CSF did not (Figure 5C). Western blotting of the BMDM pellets showed that HIF-1α protein was stabilized in conditions containing 40 µM FG-4497, whereas G-CSF was unable to stabilize HIF-1α protein. Therefore, stabilization of HIF-1α by FG-4497 is a strong stimulator of VEGF-A transcription and secretion by BMDMs in vitro, as previously shown in different malignant cell lines.23,24
To validate these results in vivo, we repeated qRT-PCR on CD11b+ macrophages, CD11b− macrophages, monocytes, granulocytes, endothelial cells, and mesenchymal progenitor cells sorted from the BM, as in Figure 3D, using Vegfa and Hprt primer–probe sets (Figure 5D). In vivo FG-4497 treatment significantly increased Vegfa mRNA in CD11b+ macrophages (Figure 5E), similar to BMDMs (Figure 5D), but not in other BM myeloid cells. Compared with saline, FG-4497 treatment also significantly increased Vegfa mRNA in Sca1+ mesenchymal progenitor cells (P = .002, Student t test), which include CXCL12-abundant reticular cells, and also caused a trend increase in more mature PDGFRα+Sca1− mesenchymal progenitor cells (P = .059). ANOVA confirmed pronounced Vegfa induction in response to G-CSF in CD11b+ macrophages, monocytes, and Sca1+ and PDGFRα+Sca1− mesenchymal progenitor cells. qRT-PCR for the 2 alternative VEGFR2 ligands Vegfc and Vegfd showed that, although these 2 cytokines were not expressed by BM myeloid cells in any condition, FG-4497 treatment increased Vegfc and Vegfd mRNA expression in Sca1+ mesenchymal progenitor cells and Vegfc expression in PDGFRα+Sca1− mesenchymal progenitor cells (Figure 5E).
We next assessed whether recombinant mouse VEGF-A164 had chemotactic effect or modulated chemotaxis of HSPCs. First, we verified that purified recombinant mouse VEGF-A was bioactive by measuring phosphorylation of VEGFR2 and Akt kinases in C166 endothelial cells (supplemental Figure 8). VEGF-A (100 ng/mL) added to the bottom chamber of a chemotaxis assay did not induce chemotaxis of HSPCs, whereas CXCL12 (50 ng/mL) did (Figure 6). To assess whether VEGF-A could modulate chemotaxis in response to the CXCL12 gradient, CXCL12 was added to the bottom chamber and VEGF-A was added to the top or bottom chamber with DC101 or control HRPN mAb. Neither VEGF-A nor DC101 mAb altered migration of mouse HPSCs in response to the CXCL12 gradient. Therefore, VEGF-A or VEGFR2 neutralization has no direct effect on HSPC chemotactic response to the CXCL12 gradient, further suggesting that the inhibitory effect of PTK787 and DC101 mAb on HSPC mobilization induced by G-CSF and FG-4497 is mediated indirectly. Considering that Sca1+ endothelial cells are the only ones that express VEGFR2 in BM tissue, these are the most likely candidates to fill this role.
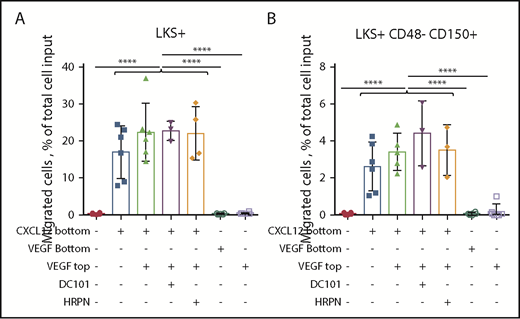
VEGF-A has no chemotactic effect on HSPCs in vitro. Chemotaxis assay with Kit+ enriched BM cells from naive C57BL/6 mice incubated for 4 hours at 37°C in the presence of 50 ng/mL CXCL12 in the bottom chamber or 100 ng/mL mouse VEGF-A in the bottom chamber or in the top insert. The percentage of LKS+ cells (A) and LKS+CD48−CD150+ HSCs (B) that migrated to the bottom chamber was measured by flow cytometry. Data are from 2 pooled experiments performed several weeks apart. Each symbol is a well. Bars are means ± standard deviation. Each individual group with CXCL12 in the bottom well was significantly different from each group without CXCL12. ****P < 10−4, ANOVA.
VEGF-A has no chemotactic effect on HSPCs in vitro. Chemotaxis assay with Kit+ enriched BM cells from naive C57BL/6 mice incubated for 4 hours at 37°C in the presence of 50 ng/mL CXCL12 in the bottom chamber or 100 ng/mL mouse VEGF-A in the bottom chamber or in the top insert. The percentage of LKS+ cells (A) and LKS+CD48−CD150+ HSCs (B) that migrated to the bottom chamber was measured by flow cytometry. Data are from 2 pooled experiments performed several weeks apart. Each symbol is a well. Bars are means ± standard deviation. Each individual group with CXCL12 in the bottom well was significantly different from each group without CXCL12. ****P < 10−4, ANOVA.
In vivo neutralization of VEGFR2 does not alter CXCL12 concentration in the BM
We next measured whether in vivo treatment with DC101 mAb altered CXCL12 concentration in the BM. G-CSF treatment for 2 days reduced CXCL12 concentration in BM fluids 2.5-fold (supplemental Figure 9), as previously reported.2 Treatment with G-CSF + FG-4497 caused a slight, but significant, additional decrease in CXCL12 concentration, in agreement with a previously reported decrease in Cxcl12 mRNA concentration.9 Surprisingly, however, in vivo neutralization of VEGFR2 with DC101 mAb did not alter CXCL12 concentration compared with mice treated with isotype control.
Discussion
With >1 million transplantations performed using mobilized blood HSPCs worldwide, this procedure has proven its benefits in improving the clinical outcome of hematological malignancies, such as acute myeloid leukemias,42,43 lymphoma,44 and myeloma.45 However, in the context of autologous transplantation, prior chemotherapy treatments and diseased BM can impair the mobilizing response to G-CSF in a high proportion of patients.1 Therefore, uncovering the cellular and molecular mechanisms of HSPC mobilization is essential to find novel treatment options to increase HSPC mobilization in these poor mobilizers, as exemplified by the recent approval of use of the CXCR4 antagonist plerixafor in combination with G-CSF for this purpose.1,2,46-49
HIF PHD inhibitors, such as FG-4497, synergize with G-CSF to enhance mouse HSPC mobilization in the C57BL/6 mouse strain,17 as well as that of human HSPCs transplanted in irradiated immune-deficient mice,26 2 models of poor mobilization.21,50 This effect is, in part, HSPC intrinsic and HIF-1 mediated, because conditional deletion of the Hif1a gene specifically in HSPCs impairs the mobilizing effect of FG-4497 on HSPCs in response to G-CSF.9 However, this does not exclude an HSPC-extrinsic effect mediated by niche cells. Herein, we show that the effect of FG-4497 on G-CSF–induced HSPC mobilization also involves stimulation of VEGFR2 signaling, because the FG-4497 effect is inhibited by PTK787, a small tyrosine kinase inhibitor selective for VEGFR1 and VEGFR2,27 as well as by DC101, a neutralizing anti-VEGFR2 mAb.
Mechanistically, we find that FG-4497 stabilizes HIF-1α protein and increases VEGF-A protein secretion by BM macrophages in vitro, consistent with the key role of BM macrophages in regulating HSC niches and promoting HSC mobilization in response to G-CSF.37,38,41,51 In vivo, FG-4497 treatment alone induced Vegfa mRNA expression in CD11b+ macrophages and Sca1+ mesenchymal progenitor cells, whereas G-CSF alone caused a stronger induction in CD11b− macrophages, monocytes, and Sca1+ and PDGFRα+ mesenchymal progenitor cells. We could not detect a significant increase in VEGF-A protein in BM fluids in response to FG-4497 in vivo. However, additional VEGF-A produced in the BM may have been consumed by increased VEGFR2 internalization following FG-4497 treatment, because VEGFR2 mRNA expression by endothelial cells was increased in response to FG-4497.
Vegfa mRNA was increased in response to G-CSF in CD11b− macrophages, monocytes, and mesenchymal progenitor cells in vivo, consistent with the presence of a STAT3 consensus binding site in human and mouse Vegfa gene promoter regions24 and the fact that G-CSF activates STAT3 phosphorylation and nuclear translocation in cells expressing its receptor.52,53 Interestingly, G-CSF was unable to stimulate VEGF-A protein production by BMDMs in vitro and CD11b+ macrophages and granulocytes in vivo. Surprisingly, we did not find a higher induction of Vegfa mRNA following G-CSF plus FG-4497 vs G-CSF alone. Interestingly, FG-4497 treatment also increased expression, at the RNA level, of VEGF-C and VEGF-D, which are also VEGFR2 ligands, albeit with a weaker affinity.29
Our observation that FG-4497, which stabilizes HIF-1α protein in the BM,9,12 increases HSPC mobilization in response to G-CSF in a VEGFR2-dependent manner is also consistent with previous reports showing that systemic elevation of circulating VEGF-A causes mobilization of CFCs and radioprotective HSCs, an effect that is blocked by neutralizing anti-VEGFR2 mAb DC101.26 Interestingly, we find that in vivo neutralization of VEGFR2 by DC101 mAb caused a dramatic increase in VEGF-A concentration in blood and BM fluids, suggesting that most VEGF-A consumption is caused by VEGFR2-mediated internalization by endothelial cells in vivo or that a compensation feedback due to VEGFR2 signaling blockade takes place, leading to enhanced VEGF-A production. Therefore, it remains possible that our measurement of VEGF-A protein concentration in BM fluids underestimates the increase in VEGF-A bioavailability and signaling in the BM in response to G-CSF and FG-4497, because of its high VEGFR2-mediated turnover, and that VEGF-C and VEGF-D also contribute to enhanced HSPC mobilization in response to FG-4497.
Although cell surface expression of VEGFR2 has been reported in mouse hematopoietic precursor cells derived from embryoid bodies,54 the expression of VEGFR1 and VEGFR2 on adult mouse BM HSPCs remains controversial. VEGFR1 was reported on adult BM LKS+ cells, with VEGFR1+ BM mononuclear cells providing radioprotection.55 VEGFR2 was also reported to be expressed by radioprotective BM Lin−Kit+ cells.56 However, in a subsequent study, VEGFR2 could not be detected at the surface of adult LKS+CD34− Hoechst33342 side population cells, and sorted VEGFR2+ cells could not reconstitute hematopoiesis in transplantation assays.57 To further assess VEGFR1 and VEGFR2 expression status in adult mouse BM HSPCs, we examined their expression at the mRNA level by performing RNA-seq on sorted LKS+ cells, as well as in silico analyses using a publicly available single-cell RNA-seq database31 and the Gene Expression Commons database.30 We also performed multicolor flow cytometry with directly conjugated mAbs specific for VEGFR1 and VEGFR2 that we validated on the mouse embryonic endothelial cell line C166 (positive for both) and the mouse mesenchymal progenitor cell line Kusa4b10 (negative for both). VEGFR1 and VEGFR2 mRNA could not be detected in any population of Lin−Kit+Sca1+ HSPCs in the BM in steady-state or in response to G-CSF. Furthermore, in our flow cytometry analyses, VEGFR2 was not detected at the cell surface of BM HSPCs, myeloid cells, or mesenchymal cells but was exclusively expressed by Sca1+ endothelial cells. Likewise Kdr (VEGFR2) mRNA could only be detected in sorted BM endothelial cells by qRT-PCR. This suggests that VEGFR2 signaling enhances HSPC mobilization in response to FG-4497 indirectly via BM endothelial cells expressing VEGFR2. In support of this, VEGF-A protein or DC101 mAb had no direct effect on LKS+ cell chemotaxis in vitro. Our findings in the adult mouse are consistent with the lack of expression of FLT1 and KDR mRNA on human BM CD34+Kit+Rhodamine123low HSPCs and the lack of proliferative effect of VEGF-A on these cells.58
The mechanism by which VEGFR2 signaling enhances HSPC mobilization may involve vascular remodeling in the BM.26,59 Indeed, although FG-4497 administration, together with G-CSF, further decreased CXCL12 in the BM compared with mice treated with G-CSF alone, this additional decrease in CXCL12 concentration was relatively modest compared with the decrease induced by G-CSF treatment; therefore, it is unlikely to explain the enhanced HSPC mobilization in response to FG-4497. On the other hand, expression of VEGF-A by tumor cells has been shown to cause vascular dilation, increased vascular density, and permeability to high-molecular-weight dextran, causing leukocyte mobilization into the circulation.59 This effect was not observed in mice bearing tumors overexpressing placental growth factor or VEGF-B, which both bind exclusively to VEGFR1.59 Furthermore, VEGF-A’s effect on BM vascular dilation and BM leukocyte mobilization was blocked by DC101 mAb or by conditional deletion of the Kdr gene in endothelial cells, but not by a neutralizing anti-mouse VEGFR1 mAb,59 demonstrating that the effect of VEGF-A on BM vascularization is solely mediated by VEGFR2 on endothelial cells.59 Finally, the promobilizing effect of vascular dilation has been documented recently by showing that PDE5 inhibitor Viagra increases HSPC mobilization in response to CXCR4 antagonist AMD3100,60 similar to the effect of FG-4497 on plerixafor-induced HSPC mobilization.9
In conclusion, we propose that PHD inhibitors increase HSPC mobilization in response to G-CSF and CXCR4 antagonist AMD31009 by 2 distinct mechanisms: (1) HSPC-intrinsic mechanisms by which PHD inhibitors further stabilize HIF-1 protein in HSPCs, enabling transcription of intracellular factors perturbing CXCR4 internalization, which decreases HSPC chemotactic response to CXCL12 and facilitates HSPC migration out of the BM9 in response to the attenuation of the CXCL12 gradient that takes place following treatment with G-CSF2,61,62 or AMD3100,63,64 and (2) an HPSC-extrinsic mechanism involving increased expression of VEGFR2 ligands VEGF-A, VEGF-C, and VEGF-D in the BM, leading to increased VEGFR2 signaling in BM endothelial cells.
The full-text version of this article contains a data supplement.
Acknowledgments
This work was supported by project grant APP1061333 from the National Health and Medical Research Council of Australia (NHMRC) (J.-P.L. and A.C.P.) and by funds from the Mater Foundation. I.G.W. and J.-P.L. are supported by Senior Research Fellowships (APP1108352 and APP1044091) from the NHMRC. T.M. was on sabbatical leave in Australia funded by Fukuoka University.
Authorship
Contribution: K.B., M.E.B., T.M., G.M. and I.G.W. coordinated the work, planned and performed experiments, interpreted results, and edited the manuscript; C.M., B.N., W.F., and T.K. performed experiments; J.D. provided expertise for BMDM experiments; A.C.P. supervised RNA-seq experiments; G.W. and L.F. provided FG-4497 and edited the manuscript; and J.-P.L. conceived the work, planned and performed experiments, interpreted results, and wrote and edited the manuscript.
Conflict-of-interest disclosure: L.F. and G.W. are employees and own equity in FibroGen, Inc., which owns the commercial rights to FG-4497. The remaining authors declare no competing financial interests.
Correspondence: Jean-Pierre Levesque, Stem Cell Biology Group, Mater Research Institute–The University of Queensland, Translational Research Institute, 37 Kent St, Woolloongabba, QLD 4102, Australia; e-mail: jp.levesque@mater.uq.edu.au.

