

Key Points
pF8 mediates long-term FVIII expression by targeting liver sinusoidal endothelial cells in hemophilic mice.
FVIII expression in liver sinusoidal endothelial cells corrects bleeding phenotype preventing inhibitor formation.

Abstract
Here we describe a successful gene therapy approach for hemophilia A (HA), using the natural F8 promoter (pF8) to direct gene replacement to factor VIII (FVIII)–secreting cells. The promoter sequence and the regulatory elements involved in the modulation of F8 expression are still poorly characterized and biased by the historical assumption that FVIII expression is mainly in hepatocytes. Bioinformatic analyses have highlighted an underestimated complexity in gene expression at this locus, suggesting an activation of pF8 in more cell types than those previously expected. C57Bl/6 mice injected with a lentiviral vector expressing green fluorescent protein (GFP) under the pF8 (lentiviral vector [LV].pF8.GFP) confirm the predominant GFP expression in liver sinusoidal endothelial cells, with a few positive cells detectable also in hematopoietic organs. Therapeutic gene delivery (LV.pF8.FVIII) in hemophilic C57/Bl6 and 129-Bl6 mice successfully corrected the bleeding phenotype, rescuing up to 25% FVIII activity, using a codon-optimized FVIII, with sustained activity for the duration of the experiment (1 year) without inhibitor formation. Of note, LV.pF8.FVIII delivery in FVIII-immunized HA mice resulted in the complete reversion of the inhibitor titer with the recovery of therapeutic FVIII activity. Depletion of regulatory T cells (Tregs) in LV-treated mice allowed the formation of anti-FVIII antibodies, indicating a role for Tregs in immune tolerance induction. The significant blood loss reduction observed in all LV.pF8.FVIII-treated mice 1 year after injection confirmed the achievement of a long-term phenotypic correction. Altogether, our results highlight the potency of pF8-driven transgene expression to correct the bleeding phenotype in HA, as well as potentially in other diseases in which an endothelial-specific expression is required.
Introduction
Hemophilia A (HA) is a rare X-linked recessive bleeding disorder characterized by either the lack of or the reduced activity of coagulation factor VIII (FVIII).1,2 Although several therapeutic options have been developed or are becoming available for HA treatment, they are still ineffective in preventing inhibitor formation.3,4 Gene therapy could offer a definitive cure for HA, liberating individuals from the need for regular intravenous delivery and limiting the immunogenicity related to direct protein infusion.5-8 Because an orthotropic liver transplantation corrected HA, the liver has been considered the primary site of FVIII production9-12 and hepatocytes in the primary FVIII-expressing cells.13-16 Over the course of recent years, however, this observation has been partially revisited because of the demonstration of FVIII synthesis and secretion by liver sinusoidal endothelial cells (LSECs; lymphatic endothelial and hematopoietic cells), rather than hepatocytes.17-30 At this time, liver-directed gene therapy for the treatment of hemophilia is among the most successful applications of gene therapy.31,32 Ongoing gene therapy trials for HA are being performed using nonintegrating AAV vectors expressing FVIII codon-optimized enhanced forms under the control of hepatocyte-specific promoters.33,34 We recently reported that the use of cell type-specific promoters to target FVIII expression in endothelial (vascular endothelial–cadherin promoter) or myeloid cells (CD11b promoter) resulted in long-term FVIII activity with no immune responses in HA mice.35 Although being effective in mice, and potentially curative, vascular endothelial–cadherin and CD11b promoters force the expression of the transgene under nonphysiological settings. The reestablishment of healthy protein expression in its physiological cells and at physiological levels, with the endogenous complexity of regulatory networks, could represent an alternative and more potent strategy for gene therapy. In this context, the F8 promoter (pF8) itself represents the ideal candidate. Initial studies investigating the F8 gene described pF8 as an 1175-bp region upstream of the translation start site and containing a transcription start site at −170 bp (NM_000132).36 Previous studies have been biased by the past assumption that FVIII is principally expressed by hepatocytes, and as such, they did not consider or analyze the promoter activity in other cell types.37,38 In addition, no study has been performed to test the ability and target cell specificity of this promoter region in driving FVIII expression in vivo. Thus, the pF8 region has remained poorly characterized. In the present study, we have investigated the ability of pF8 to drive transgene expression (green fluorescent protein [GFP] and FVIII) in a lentiviral vector (LV) construct in vivo. Our study aims to unveil pF8 activity and its cell specificity, testing its possible application in gene therapy approaches for HA and other diseases in which an endothelial-specific transgene expression is required.
Methods
FANTOM5 data analysis and data access
Analysis of the FANTOM5 collection of human libraries was performed using the Zenbu browser genomic tool39 and publicly available FANTOM5 data sets (http://fantom.gsc.riken.jp/5/).40-42 To annotate transcription start sites (TSSs) across the genome, the FANTOM5 consortium developed a decomposition-based peak identification method. Highly reliable TSSs were defined as “robust” and were obtained by applying tag evidence thresholds. For the F8 analyses, we focused our attention exclusively on the robust set of TSSs. Genomic coordinates of human F8 TSSs were extracted from Zenbu. Quantitation of F8 expression from each TSS is based on Cap Analysis of Gene Expression reads and normalization, using the relative log expression method.40 Data are expressed as tags per million and filtered with a cutoff of 3 tags/library. Expression values were extracted with specific tools available from the web-based Zenbu interface.
Transcription factor binding site prediction on pF8
In silico analysis of pF8 sequence was performed using PROMO 3.0, a web-based tool for the prediction of transcription factor binding sites (TFBS).43,44 The dissimilarity rate between the target sequence on pF8 and a specific TF consensus sequence was set up for less than 5%. For each TF, 2 parameters were considered: the number of recognized nucleotides and the number of consensus sequences on the pF8 sequence.
pF8 cloning in lentiviral vector transfer construct
The pF8 region (1175 bp) was amplified by polymerase chain reaction from human genomic DNA and inserted into LV. To obtain LVs expressing human FVIII-BDD (FVIII), FVIII.N6 (kindly provided by S. Pipe), FVIII.RH, and a codon-optimized FVIII (FVIII.co), cDNAs were inserted in the LV.pF8.GFP by substituting the GFP transgene. Primers used for cloning and sequencing are reported in supplemental Table 1.
Lentiviral vector production
Third-generation LVs were produced according to published protocols.45 Collected vector particles were titrated after transduction of ECV-304 or 293T by fluorescence-activated cell sorter (FACS) for GFP constructs or as number of integrated LV copies by quantitative polymerase chain reaction for the FVIII constructs. Primers used for quantitative polymerase chain reaction are listed in supplemental Table 1.
Animals
Animal studies were carried out according to an approved protocol by the Animal Care and Use Committees of Università del Piemonte Orientale. In vivo experiments were performed on 8- to 10-week-old mice. For GFP expression studies, LVs were delivered to C57Bl/6 wild-type mice. C57Bl/6-HA mice were used for in vivo and ex vivo gene therapy studies, with LVs expressing FVIII, FVIII.N6, and FVIII.RH. Bl/6-129-HA mice were used to evaluate in vivo FVIII activity after gene delivery, using LV.pF8.FVIII and LV.pF8.FVIII.co. For immunization, mice were subcutaneously injected with 4 U recombinant B domain-deleted hFVIII (Refacto, Pfizer) in incomplete Freund’s adjuvant. Immunocompromised nonobese diabetic/severe combined immunodeficiency-γNull HA mice (NSG-HA)30 used for hematopoietic stem cell (HSC) transplantation studies were sublethally conditioned with 50 mg/kg busulfan injected 24 to 26 hours before transplantation. NSG-HA mice were kept in autoclaved microisolator cages and fed with sterile food and water. Immunocompetent HA mice were lethally conditioned by intraperitoneal busulfan injections (25 mg/kg) from day −4 to day −1 before transplantation.46
Mouse and human hematopoietic stem cell isolation and transplantation
To isolate murine HSC (lineage negative cells, Lin−), the bone marrow (BM) was flushed from femurs, tibiae, and humeri of 6- to 8-week-old mice. Lin− cells were obtained by immunomagnetic negative selection from total BM cells, using Lineage Cell Depletion Kit (Miltenyi Biotec), transduced with LVs at a multiplicity of infection (MOI) 100 and cultured at a density of 1 × 106/mL in STEM-SPAM medium (Lonza). Human (h)CD34+ was isolated from cord blood, as previously described,30 and cultured at a density of 1 × 106/mL in STEM-SPAM medium combined with 5 ng/mL human thrombopoietin, 50 ng/mL human stem cell factor, 50 ng/mL h-interleukin 3, and 50 ng/mL human FMS-like tyrosine kinase 3 ligand. hCD34+ were transduced with LVs at an MOI 30. Twenty-four hours after transduction, 3 to 6 × 105 hCD34+ and 106 Lin− cells were suspended in STEM-SPAM and tail vein injected into the mice.
Analysis of blood and organs of transplanted mice
The engraftment of transplanted mice was measured as the percentage of GFP+ or hCD45+ cells in the peripheral blood (PB) and assessed every 4 weeks for up to 3 months by collecting PB with a retroorbital puncture. To assess the engraftment, total white cells were analyzed by flow cytometry for GFP or incubated with anti-human CD45 PE-conjugated antibody. Splenocytes were obtained after spleen digestion for 30 minutes at 37°C in Hanks balanced salt solution (Sigma-Aldrich) containing 0.2 mg/mL collagenase IV (Gibco) and filtered through a 70-µm cell strainer (Falcon). For engraftment analyses, BM cells were obtained by flushing femurs, whereas hepatocytes and liver nonparenchymal cells (NPCs) were isolated after liver perfusion.20
Immunofluorescence
The liver and spleen from the injected mice were fixed in 4% paraformaldehyde, equilibrated in 30% sucrose, and embedded in optimal cutting temperature medium. Five 6-µm cryostat sections were blocked in phosphate-buffered saline (PBS) containing 5% goat serum, 1% bovine serum albumin, 0.1% Triton X-100 (PBS-T). Primary antibodies were diluted in PBS-T. After washing, samples were incubated with secondary antibodies and 4′,6-diamidino-2-phenylindole in PBS containing 1% bovine serum albumin, 0.1% Triton X-100, and mounted with Mowiol (Sigma-Aldrich). Primary and secondary antibodies used are listed in supplemental Tables 3 and 4.
FVIII activity assays and anti-FVIII antibodies detection
FVIII activity on plasma of treated mice was measured using the activated partial thromboplastin time (aPTT) and tail clip assays. Plasma was obtained by collecting PB in 3.2% citrate and centrifuging at 2000g for 15 minutes. Standard curves were generated by serially diluting Refacto in pooled hemophilic mouse plasma. Results were expressed as a percentage of correction. The tail clip assay was performed with some modifications of a previously described protocol.47 We cut off the entire distal portion of the tail (diameter, 2-2.5 mm) of anesthetized mice, blood was collected for 10 minutes in 14 mL saline solution at 37°C, and bleeding times were recorded. Precipitated red blood cells were treated with red blood lysis buffer (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA) and the absorbance measured at 575 nm, using a VictorX (PerkinElmer) spectrophotometer. Results were analyzed by comparing the amount of blood lost from LV-corrected HA mice with that of wild-type and untreated mice.
Enzyme-linked immunosorbent assay (ELISA) on plasma from LV-injected mice to evaluate the presence of anti-FVIII antibodies has been previously described.35
Statistical analysis
The statistical analyses were performed with GraphPad Prism 5.0 (GraphPad Software). All data are expressed as an average ± standard deviation. A 1-way analysis of variance with a post hoc Bonferroni’s test was performed to compare the HA phenotypic correction between groups. A 2-way analysis of variance was used to resolve the overall effects between the different mice groups over time. Where the 2-way analysis of variance was significant, a Bonferroni’s multiple comparison test was used. Statistical significance was assumed for P < .05.
Results
F8 promoter expression atlas
We used FANTOM5 data resources to dissect and confirm TSSs involved in F8 expression and to accurately map alternative promoter regions across approximately 2000 samples of human origin. Using the FANTOM5 Zenbu Genome Browser web tool,39 we found that 5 independent TSSs are associated with human F8 (Figure 1A). TSS1 (hg19, chrX:154250990-154251004) corresponds to the region already described,36-38 which promotes the transcription of the annotated reference sequence (NM_000132). TSS1 resulted in being the most frequently used in FANTOM5 samples. In human tissues, including those relevant to HA, expression of F8 could be detected in the liver, spleen, lymph nodes, and heart (30-50 tags per million; Figure 1B). Very low to undetectable expression levels were detected in the brain. Unexpectedly, FANTOM5 analysis also highlighted a strong usage of TSS1 in adipose tissue (72.23 tags per million). In addition to the described F8 TSS1 and reference sequence, our analysis of FANTOM5 data identified novel TSSs associated to F8 expression; specifically, TSS2 (hg19, chrX:154255207-154255230), TSS3 (hg19,chrX:154251026-154251031), TSS4 (hg19, chrX:154251772-154251775), and TSS5 (hg19, chrX:154256126-154256129), thus supporting the assumption of an underestimated complexity in gene expression at this locus. On the basis of these observations, we focused our attention on the promoter covering TSS1 known as pF8 and validated by FANTOM5 analysis.
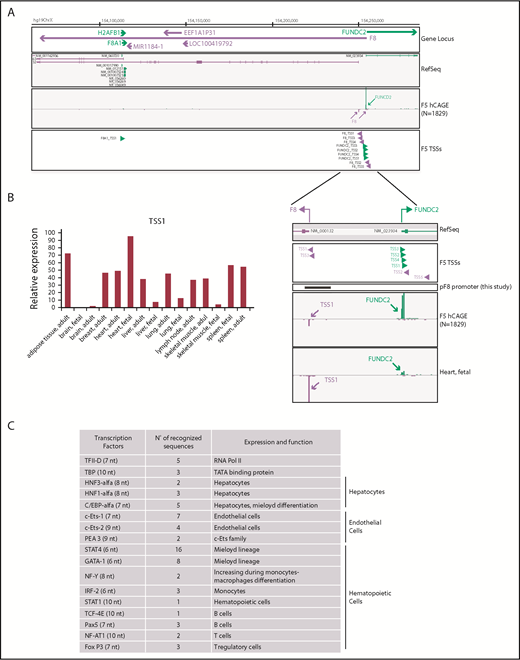
Identification of putative and alternative TSSs in human F8 gene. (A) Zenbu Genome Browser view of human F8 gene locus. Genomic coordinates in hg19 chromosome X are shown on top. Genes and transcripts are color-coded according to their orientation in the genome (positive strand, green; negative strand, purple). A track with annotated reference sequence for each gene at this locus is included (RefSeq). Expression across the whole collection of FANTOM5 human libraries is shown (F5 human Cap Analysis of Gene Expression, N = 1829). Green and purple arrowheads indicated transcription initiation in sense and antisense orientation, respectively. (B) TSS1-driven expression of F8 in human tissues (fetal, adult) measured by F5 Cap Analysis of Gene Expression analysis. (C) In silico analysis of pF8 sequence predicts the presence of several TFBS belonging to TFs expressed in hepatocytes and hematopoietic and endothelial cells. In parentheses are the numbers of nucleotides (nt) recognized.
Identification of putative and alternative TSSs in human F8 gene. (A) Zenbu Genome Browser view of human F8 gene locus. Genomic coordinates in hg19 chromosome X are shown on top. Genes and transcripts are color-coded according to their orientation in the genome (positive strand, green; negative strand, purple). A track with annotated reference sequence for each gene at this locus is included (RefSeq). Expression across the whole collection of FANTOM5 human libraries is shown (F5 human Cap Analysis of Gene Expression, N = 1829). Green and purple arrowheads indicated transcription initiation in sense and antisense orientation, respectively. (B) TSS1-driven expression of F8 in human tissues (fetal, adult) measured by F5 Cap Analysis of Gene Expression analysis. (C) In silico analysis of pF8 sequence predicts the presence of several TFBS belonging to TFs expressed in hepatocytes and hematopoietic and endothelial cells. In parentheses are the numbers of nucleotides (nt) recognized.
In silico analysis of TFBS distributed on pF8
To elucidate which TFs could be involved in FVIII expression regulation, we performed an in silico analysis of the 1175-bp pF8 sequence. This analysis, in addition to confirming the presence of TFBS belonging to HNF1, HNF3, C/EBPα, and C/EBPβ, as previously described,37,38 also revealed the presence of several TFBS potentially recognized by endothelial and hematopoietic TFs, such as Pea3, Ets-1, Ets-2, GATA1, IRF-2, STAT1, and STAT4 (Figure 1C). This prediction highlights the presence of a complex FVIII transcriptional regulation and supports the reported extrahepatic FVIII expression23-26 and production in endothelial and hematopoietic cells.9,17,18,48
In vivo pF8 activity
To evaluate in vivo pF8 activity, 5 × 108 transducing units (TU) of LV.pF8.GFP were tail vein injected into C57BL/6 mice, and the distribution of GFP was assessed by immunofluorescence (IF) and FACS analysis of liver and hematopoietic organs. The IF staining highlighted a robust GFP expression both in hepatic Lyve-1+ cells (LSECs; Figure 2A) and in splenic F4/80+ macrophages (Figure 2B) for up to 6 months. According to IF, the FACS analysis confirmed that the majority of GFP+ cells belonged to the NPC fraction (25% ± 6%; Figure 2C) and were positive for the LSEC markers Tie-2 (92% ± 4%), CD146 (93% ± 3%), and CD31 (91% ± 6%) (Figure 2D). Only less than 1% of total hepatocytes expressed GFP. FACS analysis of hematopoietic organs confirmed a predominant GFP expression in myeloid CD11b+ and CD11c+ cells, rather than B cells (B220+), T cells (CD4+ and CD8+), and granulocytes (Gr-1+; supplemental Figure 1A-B). In addition, we found rare GFP+ cells in lungs and kidneys 1 month after vector delivery. In lungs, GFP+ cells were CD31+ (ECs) but not F4/80+ (macrophages), whereas kidneys were negative for both markers (supplemental Figure 2).
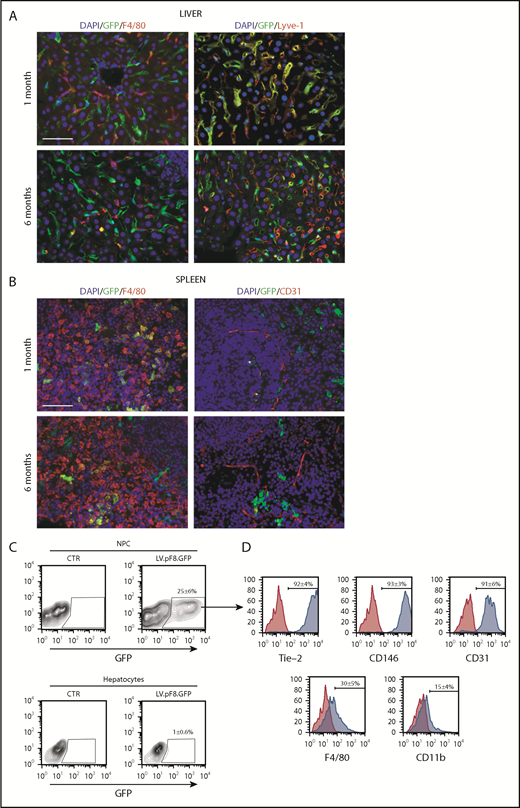
pF8 differentially drives GFP expression in liver and spleen. Representative immunofluorescence analysis of mouse liver (A) and spleen (B) of C57BL/6 HA, 1 and 6 months after LV.pF8.GFP injection. Scale bars, 50 μm. (C) Flow cytometry analysis showing GFP expression pattern in liver NPCs and hepatocytes isolated from C57BL/6 mice 1 month after LV.pF8.GFP delivery. (D) Evaluation of LSEC markers Tie-2, CD146, and CD-31 on GFP+ cells of the NPC fraction.
pF8 differentially drives GFP expression in liver and spleen. Representative immunofluorescence analysis of mouse liver (A) and spleen (B) of C57BL/6 HA, 1 and 6 months after LV.pF8.GFP injection. Scale bars, 50 μm. (C) Flow cytometry analysis showing GFP expression pattern in liver NPCs and hepatocytes isolated from C57BL/6 mice 1 month after LV.pF8.GFP delivery. (D) Evaluation of LSEC markers Tie-2, CD146, and CD-31 on GFP+ cells of the NPC fraction.
To further define the GFP expression pattern, we selectively restricted transgene expression in target cells by inserting the miRNA target (mirT) sequences of the hematopoietic-specific miRNA 142-3p (mirT-142-3p) or the endothelial miRNA 126 (mirT-126) at the 3′ of the expression cassette of LV.pF8.GFP. The presence of mirT-142-3p did not reduce the number of GFP+ cells in the liver (supplemental Figure 3A-B); as expected, however, it silenced the GFP expression in the spleen (supplemental Figure 3C). In contrast, mirT-126 caused a dramatic reduction of GFP expression in the liver (supplemental Figure 3D-E), but not in the spleen, where its expression was maintained in F4/80+ cells (supplemental Figure 3F). Taken together, the in vivo studies demonstrate that pF8 is mainly activated in ECs and in monocyte/macrophages in an organ-specific manner.
LV.pF8.FVIII delivery in hemophilic mice corrects the bleeding phenotype
To evaluate the power of pF8 to direct and rescue FVIII expression, 109 TU of LV.pF8.FVIII were injected in C57BL/6 HA mice (n = 6). Treated animals restored FVIII activity to therapeutic levels (∼9%; Figure 3A), which remained stable for up to 1 year. Interestingly, a significant improvement of FVIII activity (1.4-fold) was observed by injecting HA mice with the more active forms of FVIII, such as FVIII.RH and FVIII.N649,50 (FVIII.RH, n = 3; FVIII.N6, n = 5; Figure 3A). It should be emphasized that all treated mice did not develop inhibitors, even after immunization with FVIII (Figure 3B), whereas naive control mice developed anti-FVIII antibodies after this immunization protocol (supplemental Figure 4). A bleeding challenge performed 1 year after injection showed a significant reduction of blood loss in all LV-injected HA mice, confirming the phenotypic correction (Figure 3C-D).

In vivo gene therapy with LVs expressing FVIII and FVIII-modified forms under the control of pF8. (A) aPTT assay on plasma of C57BL/6-HA mice up to 52 weeks after LV delivery. Compared with pF8.FVIII, the injection of pF8.RH (n = 3) and pF8.N6 (n = 5) significantly improves FVIII activity 1.4-fold (∼11%) and 1.3-fold (∼10%), respectively (*P ≤ .0001). (B) ELISA assay showing no anti-FVIII antibodies over time among treated mice. Bleeding assay (C) and bleeding time (D) highlighting hemophilic phenotype correction in all FVIII-injected mice compared with HA control mice (*P ≤ .0001). Saline-injected HA mice are used as control (n = 6).
In vivo gene therapy with LVs expressing FVIII and FVIII-modified forms under the control of pF8. (A) aPTT assay on plasma of C57BL/6-HA mice up to 52 weeks after LV delivery. Compared with pF8.FVIII, the injection of pF8.RH (n = 3) and pF8.N6 (n = 5) significantly improves FVIII activity 1.4-fold (∼11%) and 1.3-fold (∼10%), respectively (*P ≤ .0001). (B) ELISA assay showing no anti-FVIII antibodies over time among treated mice. Bleeding assay (C) and bleeding time (D) highlighting hemophilic phenotype correction in all FVIII-injected mice compared with HA control mice (*P ≤ .0001). Saline-injected HA mice are used as control (n = 6).
Preexisting immunization in HA mice does not affect FVIII expression
Treatment of inhibitor-positive patients with HA remains 1 of the major obstacles to an effective therapy for HA. To explore whether pF8-mediated FVIII expression could lead to the reduction of preexisting antibodies, we immunized C57BL/6-HA mice with Refacto, and after inhibitor formation, we injected 109 TU of LV.pF8.FVIII (n = 4; Figure 4A). All immunized mice developed high titer inhibitors (≥20 BU) 4 weeks after immunization and at the time of vector administration, with a high level of immunoglobulin G1 and immunoglobulin G2a isotypes (Figure 4B-C). Interestingly, 2 weeks after vector injection, anti-FVIII antibodies began to disappear with a concomitant increase of FVIII activity (∼8%), which remained stable for up to 1 year (Figure 4A). Further, re-challenging the same mice with Refacto did not affect either FVIII activity or antibody titers (Figure 4A). Reduction of blood loss (Figure 4D) and bleeding time (Figure 4E) confirmed the phenotypic correction in all treated mice, proving that a target expression of FVIII can revert inhibitor titers and rescue FVIII activity.
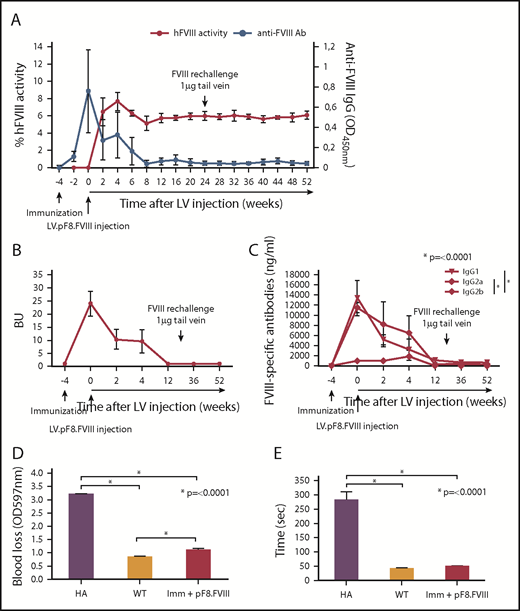
hFVIII activity after LV.pF8.FVIII delivery in HA mice previously immunized with hFVIII. (A) Graphic representation of hFVIII activity (red line) and anti-FVIII antibody (Ab) titer (blue line) trend of C57BL/6 HA injected with LV.pF8.FVIII after immunization (n = 4). (B) Bethesda assay showing formation of high titer inhibitors (≥20 BU) in all immunized mice. (C) Graph showing quantification of anti-FVIII isotypes IgG1, IgG2a, and IgG2b in plasma of FVIII-immunized and subsequently injected with LV.pF8.FVIII. High titer of IgG1 and IgG2a were predominantly detectable in all mice. Bleeding assay (D) and bleeding time (E) showing a significant phenotypic correction of HA treated mice. *P ≤ .0001.
hFVIII activity after LV.pF8.FVIII delivery in HA mice previously immunized with hFVIII. (A) Graphic representation of hFVIII activity (red line) and anti-FVIII antibody (Ab) titer (blue line) trend of C57BL/6 HA injected with LV.pF8.FVIII after immunization (n = 4). (B) Bethesda assay showing formation of high titer inhibitors (≥20 BU) in all immunized mice. (C) Graph showing quantification of anti-FVIII isotypes IgG1, IgG2a, and IgG2b in plasma of FVIII-immunized and subsequently injected with LV.pF8.FVIII. High titer of IgG1 and IgG2a were predominantly detectable in all mice. Bleeding assay (D) and bleeding time (E) showing a significant phenotypic correction of HA treated mice. *P ≤ .0001.
FVIII and codon optimized FVIII expression in B6/129 HA mice
We subsequently investigated whether LV.pF8.FVIII injection could result in a bleeding correction for a different mouse strain by delivering 109 TU of LV.pF8.FVIII in B6/129-HA mice (n = 8). The LV administration resulted in a therapeutic (∼12%) and long-lasting FVIII activity (Figure 5A), with no inhibitor development for up to 68 weeks (Figure 5B). Interestingly, the injection of 109 TU of a LV expressing a codon-optimized form of FVIII (FVIII.co) in B6/129-HA (n = 7) demonstrated a remarkable improvement in FVIII recovery, which reached 25% of activity and remained stable over the course of 1 year, with no inhibitor detection (Figure 5A-B). A reduced blood loss and shortened bleeding time validated the possibility of obtaining a complete phenotypic correction independent of the mouse strain used (Figure 5C-D). Overall, these data highlight an appreciable improvement in FVIII activity and a lack of immunogenicity in HA mice treated with the codon-optimized FVIII driven by its natural pF8 promoter.
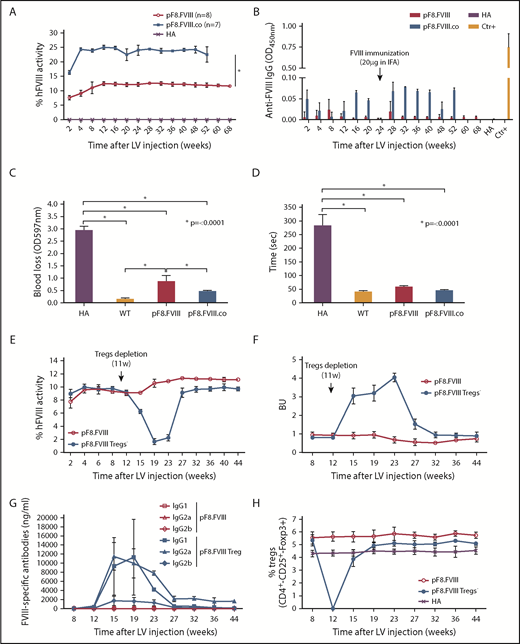
Long-term bleeding correction of B6/129-HA mice after FVIII gene transfer and effects of Tregs depletion after LV.pF8.FVIII delivery. (A) aPTT assay on plasma of LV.pF8.FVIII (n = 8) and LV.pF8.FVIII.co injected mice (n = 7) up to 68 weeks. (B) ELISA assay showing the absence of anti-FVIII antibodies after LV treatment. Bleeding assay (C) and bleeding time (D) demonstrating bleeding correction in all HA injected mice (P < .001), with a significant improvement of the HA phenotype mediated by FVIII.co form (P < .001). (E) hFVIII activity trend in plasma of B6/129-HA mice injected with LV.pF8.hFVIII (n = 10) and underwent Tregs depletion 11 weeks later (n = 5). Bethesda assay showing low titer inhibitors (F) and FVIII-specific isotypes IgG1, IgG2a, and IgG2b (G) in plasma of Tregs depleted mice. (H) Percentage of Tregs (CD4+ CD25+ Foxp3+) in PB of mice from both groups. *P ≤ .0001.
Long-term bleeding correction of B6/129-HA mice after FVIII gene transfer and effects of Tregs depletion after LV.pF8.FVIII delivery. (A) aPTT assay on plasma of LV.pF8.FVIII (n = 8) and LV.pF8.FVIII.co injected mice (n = 7) up to 68 weeks. (B) ELISA assay showing the absence of anti-FVIII antibodies after LV treatment. Bleeding assay (C) and bleeding time (D) demonstrating bleeding correction in all HA injected mice (P < .001), with a significant improvement of the HA phenotype mediated by FVIII.co form (P < .001). (E) hFVIII activity trend in plasma of B6/129-HA mice injected with LV.pF8.hFVIII (n = 10) and underwent Tregs depletion 11 weeks later (n = 5). Bethesda assay showing low titer inhibitors (F) and FVIII-specific isotypes IgG1, IgG2a, and IgG2b (G) in plasma of Tregs depleted mice. (H) Percentage of Tregs (CD4+ CD25+ Foxp3+) in PB of mice from both groups. *P ≤ .0001.
LV.pF8.FVIII expression induced Tregs, allowing transgene tolerance
To elucidate the mechanism of pF8-mediated FVIII tolerance induction, we tail vein injected 109 TU LV.pF8.FVIII into B6/129-HA mice (n = 10), and subsequently depleted the Tregs in half the group (n = 5). FVIII activity started to decrease 4 weeks after the commencement of the Tregs depletion (Figure 5E), paralleling the appearance of anti-FVIII antibodies and the eradication of the Tregs population (Figure 5E-H). Most of the detectable inhibitors were IgG1 and IgG2a isotypes, which completely disappeared after the total recovery of Tregs (Figure 5G-H). In contrast, the FVIII activity of LV.pF8.FVIII-injected control group (n = 5) remained stable (∼11%) and without the presence of inhibitors (Figure 5E-G). These data suggest that when using pF8, Tregs are required to sustain a tolerance to FVIII after an in vivo gene transfer.
pF8 activity in hematopoietic stem cell transplantation
To evaluate pF8 activity in hematopoietic cells, Lin− cells were isolated from total BM of C57Bl/6 mice, transduced with LV.pF8.GFP (pF8-Lin−) and transplanted into busulfan-conditioned C57Bl/6 mice. Lin− cells transduced with LV.PGK.GFP (PGK-Lin−) served as control. FACS analysis confirmed pF8 activity in BM-derived cells, with GFP expression detected in PB of transplanted mice with an average of 18.7% ± 6.9% for pF8-Lin− and 62.5% ± 2.1% for PGK-Lin− (supplemental Figure 5A). The GFP expression levels remained stable for up to 4 months after transplantation, demonstrated by FACS (supplemental Figure 5) and IF (Figure 6) analyses. The PB, BM, and spleen of pF8-Lin− transplanted mice revealed a marked GFP expression in myeloid cells, such as monocytes/macrophages (CD11b+), granulocytes (Gr1+), and dendritic cells (CD11c+; supplemental Figure 5B-D). In contrast, a lower but consistent GFP positivity was observed in lymphoid cells, such as B cells (CD19+) and T cells (CD4+ and CD8+ cells; supplemental Figure 5B-D) in all hematopoietic tissues analyzed. Analysis of the liver and spleen from pF8-Lin− transplanted mice revealed a predominant GFP positivity in F4/80+ macrophages (Figure 6A-B,E-F). Instead, in the spleen of PGK-Lin− transplanted mice, transgene expression was also detectable in germinal center cells in addition to the macrophages (Figure 6G-H). These data confirm that pF8 activation specifically and preferentially occurs in myeloid lineage after gene transfer and BM transplantation.
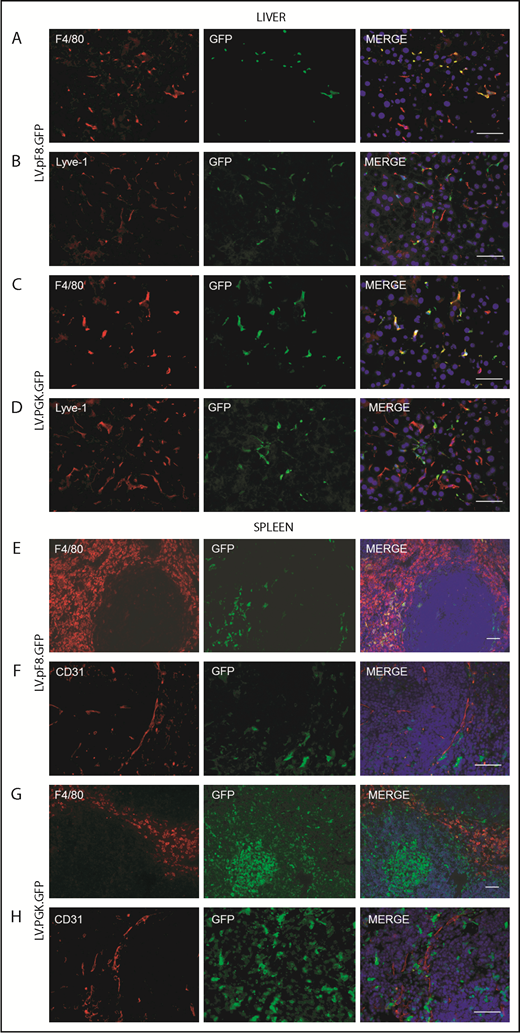
GFP expression in liver and spleen of pF8-Lin− transplanted mice. Immunofluorescence analysis of liver sections from pF8-Lin− (A-B) and PGK-Lin− C57Bl/6 transplanted mice (C-D). Immunofluorescence analysis of the spleens of pF8-Lin− (E-F) and PGK-Lin− transplanted mice (G-H). Scale bars, 50 μm.
GFP expression in liver and spleen of pF8-Lin− transplanted mice. Immunofluorescence analysis of liver sections from pF8-Lin− (A-B) and PGK-Lin− C57Bl/6 transplanted mice (C-D). Immunofluorescence analysis of the spleens of pF8-Lin− (E-F) and PGK-Lin− transplanted mice (G-H). Scale bars, 50 μm.
Ex vivo gene therapy
The combination of gene therapy and HSC transplantation has provided promising results in the treatment of HA mice and, more important, in induced immunotolerance.51-54 We opted then to investigate whether both human and mouse HSC transduced ex vivo with LV.pF8.FVIII could restore therapeutic levels of FVIII after transplantation into busulfan-conditioned HA mice. To this end, CD34+ cells, isolated from human cord blood, were transduced with LV.pF8.FVIII at MOI 30, whereas Lin− cells, isolated from BM of C57BL/6-HA mice, were transduced with MOI 100. Vector copy number quantification showed an average of 4 LV copies/cell for hCD34+ cells and 3 for Lin− cells. Transduced hCD34+ (6 × 105) were intravenously injected into NSG-HA mice (n = 8), whereas mice transplanted with untransduced hCD34+ cells served as the control group (n = 4). For murine cells, 106 LV.pF8.FVIII-transduced Lin− cells were transplanted into C57BL/6-HA mice (n = 4). FACS analysis for hCD45+ cells in PB of hCD34+ cells-transplanted NSG-HA mice showed ∼30% chimerism for up to 4 months (Figure 7A). Mice transplanted with LV-transduced hCD34+ showed therapeutic levels of FVIII activity (∼10%), whereas transplantation of untransduced CD34+ cells resulted in only 2% of FVIII activity, confirming our previous data30 (Figure 7B). The bleeding assay further confirmed a phenotypic correction in all mice transplanted with LV-transduced hCD34+ cells (Figure 7C). Similar results were obtained by transplanting mice with LV-transduced murine Lin− cells, where FVIII activity reached 5% to 6% for up to 3 months, at which point they began to decrease but remained detectable for up to 6 months (Figure 7D). Anti-FVIII antibodies were never detected (Figure 7E), and a bleeding challenge definitively confirmed a phenotypic correction in all transplanted mice (Figure 7F).
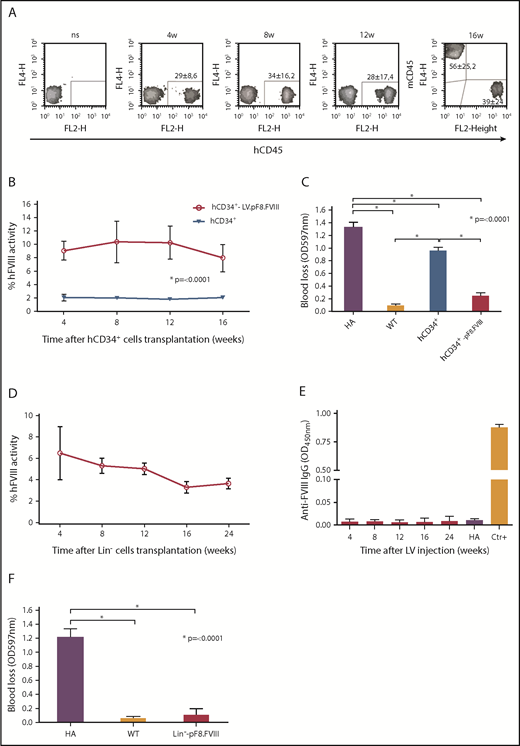
Ex vivo gene therapy using LV.pF8.FVIII. (A) FACS analysis for hD45 showing human chimerism after LV.pF8.FVIII-CD34+ transplant in NSG-HA mice for up to 3 months. aPTT (B) and bleeding (C) assays confirm therapeutic activity of FVIII and phenotypic correction in mice transplanted with LV.pF8.FVIII-hCD34+. (D) aPTT showing FVIII activity rescue after Lin− transduced cell transplantation. (E) ELISA assay demonstrates the absence of anti-FVIII antibodies in Lin− transplanted mice. (F) Bleeding assay in mice transplanted with transduced hCD34+ or murine Lin− cells demonstrate the achievement of phenotypic correction in all treated mice. *P ≤ .0001.
Ex vivo gene therapy using LV.pF8.FVIII. (A) FACS analysis for hD45 showing human chimerism after LV.pF8.FVIII-CD34+ transplant in NSG-HA mice for up to 3 months. aPTT (B) and bleeding (C) assays confirm therapeutic activity of FVIII and phenotypic correction in mice transplanted with LV.pF8.FVIII-hCD34+. (D) aPTT showing FVIII activity rescue after Lin− transduced cell transplantation. (E) ELISA assay demonstrates the absence of anti-FVIII antibodies in Lin− transplanted mice. (F) Bleeding assay in mice transplanted with transduced hCD34+ or murine Lin− cells demonstrate the achievement of phenotypic correction in all treated mice. *P ≤ .0001.
Discussion
Gene therapy is rapidly gaining a renewed interest and is emerging as a treatment option for several diseases, as highlighted by the recent market authorization of numerous gene therapy products.31 At present, several clinical trials for HA using AAV vectors are successfully ongoing in adult patients.33 Despite this success, however, AAV vectors do not actively integrate into the host cell genome, and are lost on cell division during physiological liver growth or in case of liver disease, thus limiting their use in pediatric patients and questioning their lifelong maintenance.31,55 The use of HIV-derived LVs that stably integrate into target cells could extend the application of gene therapy to pediatric patients, overcoming the issue of lifelong expression. Although the liver, and in particular LSECs, are considered the main source of FVIII,19,56,57 its production in other sites, such as the spleen, has been described in recent years,23,25-28 and as such, the presence of unknown cell types secreting FVIII cannot be excluded. Here, the choice of pF8 aimed to extend and evaluate FVIII expression in all naturally secreting cells, thus offering the possibility of obtaining a sustained transgene expression within a therapeutic range. In the present study, the in vivo delivery of LV.pF8.GFP highlighted a long-term and organ-dependent GFP expression in which the majority of GFP+ cells were represented by LSECs in the liver and myeloid cells in the spleen. Transplantation of Lin− LV.pF8.GFP-transduced cells further confirmed the activation of pF8 in myeloid lineages that resulted in being the predominant GFP+ population detected. These data, combined with the in silico prediction of TFBS distributed across pF8 genomic region, strengthened the evidence of a preferential pF8 activation in endothelial and hematopoietic cells, rather than in hepatocytes.22,30,58 Interestingly, the LV delivery of a functional copy of FVIII in HA mice restored therapeutic FVIII activity levels (∼12%), which remained stable for up to 16 months without inhibitor formation. We therefore speculate that the rescue of FVIII activity observed here, was a result of the activation of the promoter, not only in its native producing cells, the LSECs but also in others such as the hematopoietic lineage. As described, by delivering optimized forms of FVIII, such as FVIII.RH, FVIII.N6, and FVIII.co,50,59,60 we significantly improved FVIII activity up to 25%. We should emphasize that codon-optimization despite increasing protein production is a complex manipulation that may have unexpected adverse effects. Synonymous changes in the native protein sequence could alter its conformation and stability, modify the sites of posttranslational modifications, or lead to alternative peptides with unknown biological activities. In addition, there is an increased risk of developing antibodies to the exogenous protein.61 It should be highlighted that in our experiments, the use of optimized FVIII significantly improved protein activity without influencing the immune response. In addition, we were able to revert inhibitor titers and restore FVIII activity in HA mice previously immunized with FVIII. The Tregs depletion after FVIII gene transfer in HA mice induced inhibitor formation with the consequent disappearance of FVIII activity. Interestingly, this effect is reversed as soon as new Tregs are produced and detected in the periphery. These results, which confirm our previous findings on FVIII gene therapy, using an endothelial-specific promoter,35 support the hypothesis that LSECs could act as tolerogenic antigen-presenting cells recruiting Tregs and preventing the enrollment of activating antigen-presenting cells.62 Finally, ex vivo gene therapy using LV.pF8.FVIII to transduce both murine and human HSC resulted in FVIII levels around 10% in HA-transplanted mice. The different FVIII activity achieved by transplanting hCD34+ rather than murine Lin− could be explained by the human origin of the promoter that works better in hCD34+-derived transplanted cells and by the higher transduction capacity of LVs in human cells. Taken together, these studies demonstrate that pF8 is suitable for HA gene therapy because of its ability to drive therapeutic FVIII expression and to prevent inhibitor formation by targeting LSECs and splenic macrophages.
Because of the complex nature of FVIII expression, further studies on pF8 are necessary to gain insights into FVIII transcriptional regulation in different organs, evaluating the role of each TFBS identified in promoting pF8 activation. Based on the results obtained using the FANTOM5 data set, it is likely that additional regulatory elements are involved in controlling F8 expression from alternative promoters, especially those more distal to the cassette used in the present study (TSS2, TSS5). In addition, we speculate that these newly identified TSS could give rise to novel FVIII isoforms whose function or functions and origin or origins could be important for FVIII activity. Taken together, the FANTOM5 data strongly suggest that pF8 is active in more cell types than those previously expected, and that alternative promoters likely exist that regulate FVIII expression specificity. Finally, understanding the biological and molecular mechanisms regulating the immune-modulatory activity are crucial for the development of new strategies to prevent undesired immune reactions not only for the treatment of HA but also for other illnesses, such as autoimmune diseases.
Acknowledgments
The authors thank L. Salmi and S. Buzzi for help with some experiments; S. Pignani for help with the visual abstract; and S. Pipe, V. Arruda, and T. Vandendriesscche for providing optimized FVIII constructs. The authors thank G. Walker and C. Borsotti for English revision and critical reading of the manuscript.
This work was supported by ERC #261178 and Horizon 2020 (hemAcure project #66742) (A.F.).
Authorship
Contribution: S.M., R.F., E.B., and D.Z. designed and performed experiments and analyzed data; V.B. produced lentiviral vectors for in vivo experiments; S.Z. performed bioinformatics analyses; A.F. conceived the study, generated funding, designed the experiments, and analyzed data; and all authors wrote and revised the paper.
Conflict-of-interest disclosure: A.F., R.F., S.M., and D.Z. are named inventors of the patent “Promotore per Espressione Genica Cellulo-Specifica E Relativi Usi” (n. 102016000059985, released by the Italian Patent Office on 17 December 2018). The remaining authors declare no competing financial interests.
Correspondence: Antonia Follenzi, Department of Health Sciences, Università del Piemonte Orientale “A. Avogadro,” Via Paolo Solaroli 17, 28100 Novara, Italy; e-mail: antonia.follenzi@med.uniupo.it.

