

Key Points
Procoagulant platelets exhibit a distinct pattern of Ca2+ signaling that we term “supramaximal” Ca2+ signaling.
Our model provides a new framework to interpret the role of different platelet signaling systems in the generation of procoagulant platelets.

Abstract
Procoagulant platelets promote thrombin generation during thrombosis. Platelets become procoagulant in an all-or-nothing manner. We investigated how distinct Ca2+ signaling between platelet subpopulations commits some platelets to become procoagulant, using the high-affinity Ca2+ indicator Fluo-4, which may become saturated during platelet stimulation, or low-affinity Fluo-5N, which reports only very high cytosolic Ca2+ concentrations. All activated platelets had high Fluo-4 fluorescence. However, in Fluo-5N–loaded platelets, only the procoagulant platelets had high fluorescence, indicating very high cytosolic Ca2+. This finding indicates a novel, “supramaximal” Ca2+ signal in procoagulant platelets (ie, much higher than normally considered maximal). Supramaximal Ca2+ signaling and the percentage of procoagulant platelets were inhibited by cyclosporin A, a mitochondrial permeability transition pore blocker, and Ru360, an inhibitor of the mitochondrial Ca2+ uniporter, with no effect on Fluo-4 fluorescence. In contrast, Synta-66, an Orai1 blocker, reduced Fluo-4 fluorescence but did not directly inhibit generation of the supramaximal Ca2+ signal. Our findings show a distinct pattern of Ca2+ signaling in procoagulant platelets and provide a new framework to interpret the role of platelet signaling pathways in procoagulant platelets. This requires reassessment of the role of different Ca2+ channels and may provide new targets to prevent formation of procoagulant platelets and limit thrombosis.
Introduction
Procoagulant platelets are a subpopulation of activated platelets that expose phosphatidylserine (PS), allowing a burst of thrombin generation that is responsible for producing an occlusive thrombus.1-3 Selective inhibition of procoagulant platelets is a potential antithrombotic strategy.3
Procoagulant platelets form in an all-or-nothing manner: procoagulant platelets expose PS, whereas activated but noncoagulant platelets do not.4-6 However, almost all platelets can become procoagulant if treated with a Ca2+ ionophore, and almost all platelets become activated but noncoagulant if stimulated with some platelet activators, such as the protease-activated receptor 1 agonist SFLLRN-amide.7 Individual platelets are therefore capable of forming either subpopulation, depending on the activating stimulus. During activation, differences in intracellular signaling between activated platelets may lead platelets to commit to becoming procoagulant or noncoagulant. Increased cytosolic Ca2+ concentration ([Ca2+]cyt) is required for procoagulant and noncoagulant platelet activation, but higher or more sustained increases in [Ca2+]cyt may commit some platelets to becoming procoagulant.1,8-11 However, it is currently unclear how variation in [Ca2+]cyt between platelets leads to an all-or-nothing response.
Mitochondrial permeability transition pore (mPTP) opening is also required for platelets to become procoagulant.6 Ca2+ enters mitochondria from the cytosol through the mitochondrial Ca2+ uniporter (MCU), leading to mPTP opening above a threshold of high mitochondrial Ca2+ concentration ([Ca2+]mito).6 Cyclophilin D (CypD) reduces the threshold of [Ca2+]mito for mPTP opening.12 CypD-deficient or MCU-deficient mouse platelets generate significantly fewer procoagulant platelets than wild-type platelets.6,13,14 Cyclosporin A (CsA), which inhibits CypD, and Ru360, which inhibits the MCU, also inhibit the procoagulant platelet formation.4,5,15
Two models have been proposed to explain how mPTP opening and cytosolic Ca2+ signaling interact to commit platelets to become procoagulant. Choo et al5 reported that because [Ca2+]cyt signaling was not obviously different in CypD-deficient mouse platelets, mPTP opening causes stimulated platelets to become procoagulant without further altering [Ca2+]cyt. In contrast, Panteleev et al9,16 reported that stochastic variation in [Ca2+]cyt and [Ca2+]mito between activated platelets leads to mPTP opening in some platelets, changing [Ca2+]cyt signaling from Ca2+ spikes to sustained Ca2+ signals.
The goal of the current study was to resolve these differences and propose a new model for how platelets commit to become procoagulant in an all-or-nothing manner.
Methods
Reagents
Synta-66, thapsigargin, thrombin, and fibrinogen were from MilliporeSigma. MitoTracker Deep Red FM, annexin V (AnV)–allophycocyanin (APC) conjugate, and tandem PE-Cy7–conjugated anti-CD41 antibody, Fluo-4 acetoxymethyl ester (AM), and Fluo-5N AM were from Thermo Fisher Scientific. MitoView Green was from Biotium. CsA was from Cambridge Bioscience. Ru360 was from VWR. Cross-linked collagen-related peptide (CRP-XL) was synthesized by one of the authors (J.-D.M.) according to previously published methods.17
Platelet preparation
Blood from healthy drug-free volunteers was drawn into sodium citrate (3.2% vol/vol) with approval from the Human Biology Research Ethics Committee, University of Cambridge. Volunteers had given written informed consent in accordance with the Declaration of Helsinki. Washed platelets were prepared as previously described.18 Acid citrate dextrose (85 mM tri-sodium citrate, 71 mM citric acid, and 111 mM D-glucose) was added (1:7 vol/vol) and platelet-rich plasma was separated by centrifugation (200g, 10 minutes, ambient temperature, and without any brake). Platelets were pelleted by centrifugation (600g, 10 minutes, ambient temperature) in the presence of prostaglandin E1 (100 nM) and apyrase (0.02 U/mL). Pelleted platelets were resuspended in N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES)–buffered saline supplemented with D-glucose (135 mM NaCl, 10 mM HEPES, 3 mM KCl, 1 mM MgCl2, 0.34 mM NaH2PO4, 5 mM D-glucose, pH 7.4.) and apyrase (0.02 U/mL). Platelets were rested for 30 minutes at 30°C before treatment protocols were initiated. Where indicated, platelets were loaded with Fluo-4-AM or Fluo-5N-AM (1 µM, 10 minutes, 30°C) immediately before stimulation. Under these conditions, the dyes reported cytosolic increases in Ca2+ (supplemental Figure 1). CaCl2 was added at the final concentration of 2 mM immediately before stimulation.
Confocal imaging
Platelets (108/mL) were allowed to adhere to a fibrinogen (0.1 mg/mL)-coated coverslip for 30 minutes in the copresence of 1 μM Fluo-4-AM and 20 nM MitoTracker Deep Red FM, or 1 μM Fluo-5N-AM and 20 nM MitoTracker Deep Red FM. Nonadherent platelets (and unloaded dye) were gently washed away and HEPES-buffered saline supplemented with D-glucose replaced on the adherent platelets. Fluorescence was recorded by using a Leica SP5 confocal microscope. Platelets were then stimulated with thrombin (1 U/mL) and CRP-XL (1 µg/mL) in the presence of extracellular CaCl2 (2 mM), and samples were recorded for an additional 10 minutes.
Flow cytometry
Fluo-4– or Fluo-5N–loaded platelets were pretreated with either inhibitors or their vehicle controls as indicated in the main text followed by stimulation with thrombin (1 U/mL) and CRP-XL (1 µg/mL). AnV-APC was used to detect surface PS exposure, and PE-Cy7–conjugated anti-CD41 antibody was used to positively identify platelets. Fluorescence was detected by flow cytometry (BD Accuri C6). CD41+ events were identified as platelets based on their forward and side-scatted profile, using 1 µm silica beads to define (and exclude) microparticles. For end point analysis, 10 000 platelets were collected. For real-time flow cytometry, events were collected at a slow flow rate for 10 minutes after stimulation.
Plate reader
Fluo-4– and Fluo-5N–loaded platelets were stimulated in black 96-well plates (FLUOStar Plate Reader; excitation, 485 nm; emission, 520 nm).
Estimation of [Ca2+]cyt
Data analysis
Data presented are mean ± standard error of the mean from at least 4 independent platelet preparations. Statistical tests used are described in the figure legends.
Results
Procoagulant platelets have a “supramaximal” [Ca2+]cyt signal
Although platelet Ca2+ signaling has been widely studied with high-affinity Ca2+-sensitive dyes (eg, Fluo-4, Fura-2), these may be relatively saturated during the strong stimulation required to generate procoagulant platelets.19 To investigate this theory, platelets were loaded with two Ca2+-sensitive fluorescent dyes, either Fluo-4 or Fluo-5N (in vitro Kd = 390 nM and 90 µM, respectively). Confocal microscopy imaging of Fluo-4– and Fluo-5N–loaded platelets confirmed that these indicators reported [Ca2+]cyt in platelets under our conditions (supplemental Figure 1A-B). Platelets were then stimulated with Thr/CRP-XL and observed by using real-time flow cytometry. AnV-APC binding identified procoagulant platelets. A difference in Fluo-4 fluorescence between procoagulant (AnV+) and noncoagulant (AnV–) platelets was difficult to discern because there is considerable overlap between the Fluo-4 fluorescence distributions of these 2 subpopulations (Figure 1A-B).
![Procoagulant platelets have supramaximal [Ca2+]cyt signaling that is detected by low-affinity Fluo-5N but not high-affinity Fluo-4. (A) Human platelets were loaded with Fluo-4 then treated with CsA (2 µM) or dimethyl sulfoxide (DMSO) (as control) before stimulation with Thr/CRP-XL (arrowhead) in the presence of AnV-APC. Samples were analyzed by using real-time flow cytometry. Platelets were gated on an FSC/SSC profile to exclude microparticles. Fluo-4 fluorescence is shown. Color represents event density from low (blue) to high (red). (B) A comparison of Fluo-4 fluorescence and AnV-APC fluorescence after 10 minutes of stimulation. The horizontal line defines platelets AnV+ (procoagulant) and AnV– (noncoagulant) subpopulations. The vertical line indicates the Fluo-4 fluorescence of unstimulated platelets. All activated platelets exhibit high Fluo-4 fluorescence. (C) The Fluo-4 MFI in all platelets, AnV+ platelets, and AnV– platelets. Data are mean ± standard error of the mean (n = 5; 2-way analysis of variance with the Šidák multiple comparison test). (D) Mean Fluo-4 fluorescence (MFI normalized to initial MFI; F/F0) of all platelets from real-time flow cytometry (n = 5). The dotted line is the standard error of the mean. (E) Mean Fluo-4 fluorescence (F/F0) of platelets stimulated with Thr/CRP-XL measured in a microplate reader. (F-J) show the equivalent experiments in Fluo-5N–loaded platelets. ***P < .001 for indicated comparison; †††P < .001 for difference in MFI between AnV+ vs AnV– for DMSO or CSA, as appropriate. n.s., not significantly different for the indicated comparison.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/1/10.1182_bloodadvances.2019000182/3/m_advancesadv2019000182f1.png?Expires=1769129552&Signature=BjRzCAML-brdGYV0dB8Ay4YjouHkgaBOvfQiDJ9XC5LhjM0O0kiXV-2ea0a4Hj7OQMwegn0GOYzVczJVZPZDtG-FrY2QMy3n3Ejez39v3oRRvFjY9~Tpt1viJeQhbbjwnb9W7uXwlvWNkyV4k4770pc9cOekXCh5iREwpiKpcbnYQYbCA-sew2NRYyAFgFSmxaZajuvA0RN88mmGwGEaiESBGRd5cls~r85P8pTB~dvA4UD3~8q1ysX1zQ7a~GrWZ6alQyu5L0n7sCy9kgPy6qN5BO5bTdLYVEinuZYBK0-3cNP1zPUGnZn-zkAy4Vei~-GIQfrZ6xX3rN4rY8I5yQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Procoagulant platelets have supramaximal [Ca2+]cyt signaling that is detected by low-affinity Fluo-5N but not high-affinity Fluo-4. (A) Human platelets were loaded with Fluo-4 then treated with CsA (2 µM) or dimethyl sulfoxide (DMSO) (as control) before stimulation with Thr/CRP-XL (arrowhead) in the presence of AnV-APC. Samples were analyzed by using real-time flow cytometry. Platelets were gated on an FSC/SSC profile to exclude microparticles. Fluo-4 fluorescence is shown. Color represents event density from low (blue) to high (red). (B) A comparison of Fluo-4 fluorescence and AnV-APC fluorescence after 10 minutes of stimulation. The horizontal line defines platelets AnV+ (procoagulant) and AnV– (noncoagulant) subpopulations. The vertical line indicates the Fluo-4 fluorescence of unstimulated platelets. All activated platelets exhibit high Fluo-4 fluorescence. (C) The Fluo-4 MFI in all platelets, AnV+ platelets, and AnV– platelets. Data are mean ± standard error of the mean (n = 5; 2-way analysis of variance with the Šidák multiple comparison test). (D) Mean Fluo-4 fluorescence (MFI normalized to initial MFI; F/F0) of all platelets from real-time flow cytometry (n = 5). The dotted line is the standard error of the mean. (E) Mean Fluo-4 fluorescence (F/F0) of platelets stimulated with Thr/CRP-XL measured in a microplate reader. (F-J) show the equivalent experiments in Fluo-5N–loaded platelets. ***P < .001 for indicated comparison; †††P < .001 for difference in MFI between AnV+ vs AnV– for DMSO or CSA, as appropriate. n.s., not significantly different for the indicated comparison.
Procoagulant platelets have supramaximal [Ca2+]cyt signaling that is detected by low-affinity Fluo-5N but not high-affinity Fluo-4. (A) Human platelets were loaded with Fluo-4 then treated with CsA (2 µM) or dimethyl sulfoxide (DMSO) (as control) before stimulation with Thr/CRP-XL (arrowhead) in the presence of AnV-APC. Samples were analyzed by using real-time flow cytometry. Platelets were gated on an FSC/SSC profile to exclude microparticles. Fluo-4 fluorescence is shown. Color represents event density from low (blue) to high (red). (B) A comparison of Fluo-4 fluorescence and AnV-APC fluorescence after 10 minutes of stimulation. The horizontal line defines platelets AnV+ (procoagulant) and AnV– (noncoagulant) subpopulations. The vertical line indicates the Fluo-4 fluorescence of unstimulated platelets. All activated platelets exhibit high Fluo-4 fluorescence. (C) The Fluo-4 MFI in all platelets, AnV+ platelets, and AnV– platelets. Data are mean ± standard error of the mean (n = 5; 2-way analysis of variance with the Šidák multiple comparison test). (D) Mean Fluo-4 fluorescence (MFI normalized to initial MFI; F/F0) of all platelets from real-time flow cytometry (n = 5). The dotted line is the standard error of the mean. (E) Mean Fluo-4 fluorescence (F/F0) of platelets stimulated with Thr/CRP-XL measured in a microplate reader. (F-J) show the equivalent experiments in Fluo-5N–loaded platelets. ***P < .001 for indicated comparison; †††P < .001 for difference in MFI between AnV+ vs AnV– for DMSO or CSA, as appropriate. n.s., not significantly different for the indicated comparison.
In contrast, the difference in Fluo-5N fluorescence between the subpopulations was clear. Fluo-5N fluorescence was high in procoagulant platelets, whereas the noncoagulant platelets had fluorescence similar to unstimulated platelets (Figure 1F-G). The very low Ca2+ affinity of Fluo-5N (90 µM) means that [Ca2+]cyt in procoagulant platelets must be very high compared with noncoagulant platelets whose fluorescence was too low to be accurately reported by Fluo-5N. We term this very high Ca2+ signal in procoagulant platelets “supramaximal”; that is, much higher than normally considered maximal (1-2 µM).19
When platelets were stimulated with the Ca2+ ionophore A23187, almost all platelets became AnV+, as expected.20 These platelets also had high Fluo-4 or Fluo-5N fluorescence (supplemental Figure 2). To estimate [Ca2+]cyt in Fluo-5N–loaded procoagulant platelets, A23187 stimulation was used to estimate Fmax and BAPTA-loaded, unstimulated platelets to estimate Fmin. Another aliquot of the same preparation of Fluo-5N–loaded platelets was then stimulated with Thr/CRP-XL in the presence of AnV-APC. Using the median fluorescence intensity of procoagulant (AnV+) and noncoagulant (AnV–) platelets, [Ca2+]cyt in procoagulant platelets was estimated as 166 ± 56 µM (n = 4; see “Methods”). This is ∼1.8× the Kd of Fluo-5N and thus within the range that it is sensitive to changes in Ca2+ concentration. In contrast, it is ∼250× the Kd of Fluo-4. Although the [Ca2+]cyt in noncoagulant platelets was estimated as 3.9 ± 0.7 µM (n = 4) using Fluo-5N, we do not consider the estimate in noncoagulant platelets to be reliable, as Ca2+-sensitive dyes are likely to be inaccurate beyond 0.1 to 10× the Kd of the dye.21 This finding shows the importance of using dyes whose Ca2+ affinities are appropriate to the signal being measured.
One potential explanation for these observations is that procoagulant platelets have lost plasma membrane integrity and that Ca2+ has passively entered through this leaky plasma membrane. To test this theory, platelets were loaded with calcein-AM before stimulation. Loss of calcein fluorescence would indicate loss of plasma membrane integrity. Under these conditions, only a small proportion of procoagulant platelets lost calcein fluorescence, whereas most procoagulant platelets retained calcein (supplemental Figure 3). As a positive control, heat-killed platelets all lost calcein fluorescence. Together, these data indicate that procoagulant platelets with supramaximal Ca2+ signal have an intact plasma membrane.
Supramaximal Ca2+ signaling is dependent on mPTP opening
CsA (2 µM) significantly reduced the proportion of platelets with high Fluo-5N fluorescence (Figure 1F-G), indicating that mPTP opening is required for the supramaximal Ca2+ signal. Because the Fluo-5N fluorescence of the 2 subpopulations is very different, CsA reduced the Fluo-5N fluorescence of the whole population in flow cytometry and microplate experiments (Figure 1H-J). This outcome was not seen if Fluo-4 was used instead because the procoagulant and noncoagulant subpopulations have similar Fluo-4 fluorescence (Figure 1C-E).
To determine whether the likelihood of becoming procoagulant is related to the number of mitochondria in platelets, mitochondrial content was detected by using MitoGreen. However, there was no difference in MitoGreen fluorescence intensity between procoagulant and noncoagulant platelets (supplemental Figure 4).
A model for all-or-nothing commitment to becoming procoagulant
These data support a model in which mPTP opening commits a platelet to becoming procoagulant by triggering supramaximal [Ca2+]cyt signaling (Figure 2). Platelet activators trigger an initial increase in [Ca2+]cyt that varies between platelets, leading to interplatelet variation in [Ca2+]mito, putting some above the threshold for mPTP opening (described elsewhere16 ). mPTP opening triggers supramaximal [Ca2+]cyt signaling in these platelets, committing them to becoming procoagulant. Variation in the initial [Ca2+]cyt signal is therefore converted into 2 separate subpopulations with distinct [Ca2+]cyt signals and an all-or-nothing commitment to becoming procoagulant.
![Proposed model for the role of Ca2+signaling in the commitment to becoming procoagulant. Platelet activators trigger an initial increase in [Ca2+]cyt that varies between platelets (1). This action will lead to variation in [Ca2+]mito (2), putting some above the threshold for mPTP opening. A supramaximal [Ca2+]cyt signal is triggered in these platelets (3), committing them to becoming procoagulant. Variation in the initial [Ca2+]cyt is therefore converted into 2 distinct subpopulations with distinct [Ca2+]cyt signals, leading to an all-or-nothing commitment to becoming procoagulant.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/1/10.1182_bloodadvances.2019000182/3/m_advancesadv2019000182f2.png?Expires=1769129552&Signature=21a5CQo-5Q56shu3Oyb5iTeOE0lp9eHnBpHa3QpuIlzgoxU5mjSZZOImWNrlobeZIPCEWV63GawiCnHJDuhvICdHILXGw0HWiy49yNlH4wY6Dzevzl3Z6hgb7Jd30fcWMT5Z9zicwFVuUlM4D8RWPmxTFWoswfWXO8Ne50uzHo5-hjJVPSP~WwHL2~blKJxS2b3Bi8rSoFaaIHJe6wNFhKM0YJ~Lz9I6TV152hwPx9VRpS98P08lTLHJasdpnPGe6nN6WNXcEBkWrkTA76qsbd~A06RsMo7koFin3JSf1bwfUFFuxiYQaZXhE--GjkgOIE3w96FiKGkJ5~myxDbhnw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Proposed model for the role of Ca2+signaling in the commitment to becoming procoagulant. Platelet activators trigger an initial increase in [Ca2+]cyt that varies between platelets (1). This action will lead to variation in [Ca2+]mito (2), putting some above the threshold for mPTP opening. A supramaximal [Ca2+]cyt signal is triggered in these platelets (3), committing them to becoming procoagulant. Variation in the initial [Ca2+]cyt is therefore converted into 2 distinct subpopulations with distinct [Ca2+]cyt signals, leading to an all-or-nothing commitment to becoming procoagulant.
Proposed model for the role of Ca2+signaling in the commitment to becoming procoagulant. Platelet activators trigger an initial increase in [Ca2+]cyt that varies between platelets (1). This action will lead to variation in [Ca2+]mito (2), putting some above the threshold for mPTP opening. A supramaximal [Ca2+]cyt signal is triggered in these platelets (3), committing them to becoming procoagulant. Variation in the initial [Ca2+]cyt is therefore converted into 2 distinct subpopulations with distinct [Ca2+]cyt signals, leading to an all-or-nothing commitment to becoming procoagulant.
Reassessing the role of Ca2+ channels in procoagulant platelets
Subpopulation analysis of this all-or-nothing signaling requires reassessment of the role of different Ca2+ channels in generating procoagulant platelets. Individual Ca2+ channels could be involved in the initial [Ca2+]cyt signal, in regulation of [Ca2+]mito, or the generation of the supramaximal [Ca2+]cyt signal.
The store-operated channel, Orai1, contributes to PS exposure.22-25 However, although the Orai1 blocker Synta-66 (supplemental Figure 5) reduced the percentage of procoagulant platelets and inhibited the median Fluo-4 fluorescence in the whole platelet population (consistent with previous cuvette or microplate-based experiments24 ) (Figure 3A-D), the Fluo-5N fluorescence in platelets that had become procoagulant was unaffected (Figure 3B,E). This finding suggests that Orai1 contributes to the initial [Ca2+]cyt signal, affecting the percentage of platelets that undergo mPTP opening and become procoagulant, but is not directly involved in the generation of the supramaximal Ca2+ signal.
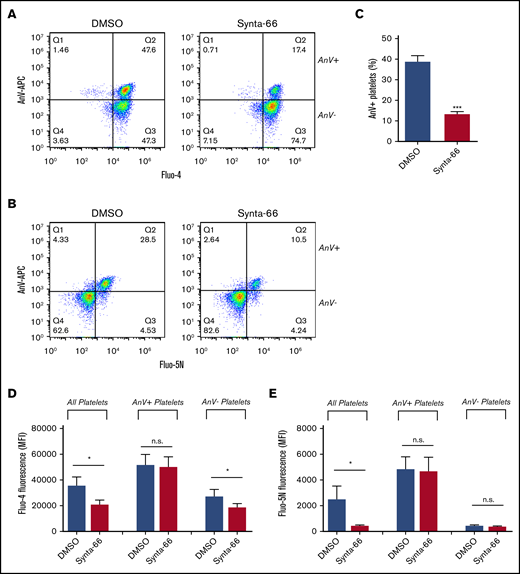
Role of SOCE in procoagulant platelet generation. To test the role of SOCE, Fluo-4– or Fluo-5N–loaded platelets were treated with the Orai1 blocker Synta-66 (10 µM), stimulated with Thr/CRP-XL in the presence of AnV-APC, and analyzed by using flow cytometry. Representative density plots are shown for Fluo-4–loaded platelets (A) or Fluo-5N–loaded platelets (B). (C) The percentage of platelets that bound AnV (***P < .001; paired Student t test). The effect of Synta-66 on Fluo-4 (D) or Fluo-5N (E) fluorescence in all platelets, AnV+, platelets and AnV– platelets. *P < .05 for dimethyl sulfoxide (DMSO) vs Synta-66–treated platelets; 2-way analysis of variance with the Šidák multiple comparison test; n = 4.
Role of SOCE in procoagulant platelet generation. To test the role of SOCE, Fluo-4– or Fluo-5N–loaded platelets were treated with the Orai1 blocker Synta-66 (10 µM), stimulated with Thr/CRP-XL in the presence of AnV-APC, and analyzed by using flow cytometry. Representative density plots are shown for Fluo-4–loaded platelets (A) or Fluo-5N–loaded platelets (B). (C) The percentage of platelets that bound AnV (***P < .001; paired Student t test). The effect of Synta-66 on Fluo-4 (D) or Fluo-5N (E) fluorescence in all platelets, AnV+, platelets and AnV– platelets. *P < .05 for dimethyl sulfoxide (DMSO) vs Synta-66–treated platelets; 2-way analysis of variance with the Šidák multiple comparison test; n = 4.
Similarly, Ru360, an inhibitor of the MCU, also reduced the percentage of procoagulant platelets (Figure 4A-C). However, Ru360 did not affect the median Fluo-4 fluorescence in the whole platelet population (Figure 4A,D), suggesting that it does not affect the initial [Ca2+]cyt signal. The Fluo-5N fluorescence of platelets that had become procoagulant was also unaffected (Figure 4B,E), suggesting that MCU is not directly involved in the generation of the supramaximal Ca2+ signal.

Role of MCU in procoagulant platelet generation. To test the role of MCU, Fluo-4– or Fluo-5N–loaded platelets were treated with the MCU blocker Ru360 (10 µM), stimulated with Thr/CRP-XL in the presence of AnV-APC, and analyzed by using flow cytometry. Representative density plots are shown for Fluo-4–loaded platelets (A) or Fluo-5N–loaded platelets (B). (C) The percentage of platelets that bound AnV (***P < .001; paired Student t test). The effect of Ru360 on Fluo-4 (D) or Fluo-5N (E) fluorescence in all platelets, AnV+ platelets, and AnV– platelets. **P < .01, ***P < .001 for dimethyl sulfoxide (DMSO) vs Ru360-treated platelets; 2-way analysis of variance with the Šidák multiple comparison test; n = 4.
Role of MCU in procoagulant platelet generation. To test the role of MCU, Fluo-4– or Fluo-5N–loaded platelets were treated with the MCU blocker Ru360 (10 µM), stimulated with Thr/CRP-XL in the presence of AnV-APC, and analyzed by using flow cytometry. Representative density plots are shown for Fluo-4–loaded platelets (A) or Fluo-5N–loaded platelets (B). (C) The percentage of platelets that bound AnV (***P < .001; paired Student t test). The effect of Ru360 on Fluo-4 (D) or Fluo-5N (E) fluorescence in all platelets, AnV+ platelets, and AnV– platelets. **P < .01, ***P < .001 for dimethyl sulfoxide (DMSO) vs Ru360-treated platelets; 2-way analysis of variance with the Šidák multiple comparison test; n = 4.
This reassessment therefore distinguishes distinct contributions of the plasma membrane channel (Orai1) and the mitochondrial Ca2+ transporters (MCU and mPTP opening) in generating procoagulant platelets.
Discussion
Procoagulant platelets form in an all-or-nothing manner, which suggests that during activation, platelets commit to becoming procoagulant or noncoagulant and that there are differences in intracellular signaling between these 2 subpopulations. Increased [Ca2+]cyt is required for both procoagulant and noncoagulant platelet activation11 ; although there is considerable variation in the [Ca2+]cyt signals between activated platelets, higher or more sustained [Ca2+]cyt signals are associated with procoagulant platelets.1,8,9,25 However, how the variation in [Ca2+]cyt signaling between activated platelets leads to an all-or-nothing phenotype is not clear.
In this study, we propose a new model to explain this. Our model (Figure 2) involves 3 different Ca2+ signaling events. First, platelet activators, such as thrombin and collagen, trigger an increase in [Ca2+]cyt that varies between platelets. This initial [Ca2+]cyt signal occurs through release of Ca2+ from intracellular Ca2+ stores and entry through plasma membrane Ca2+ channels, such as Orai1 and TRPC6.23,25 Some of the cytosolic Ca2+ is taken into mitochondria through MCU,13 increasing [Ca2+]mito. Because the initial [Ca2+]cyt signal varies between platelets, the subsequent [Ca2+]mito signal also varies. The key commitment step is then opening of mPTP, which occurs above a threshold [Ca2+]mito. This concept was first proposed by Panteleev et al in mathematical models and subsequent experiments.9,16 However, we further propose that mPTP opening triggers an additional increase in [Ca2+]cyt that is of a different order of magnitude to the initial [Ca2+]cyt signal, and much larger than any [Ca2+]cyt signal previously reported in platelets. This supramaximal [Ca2+]cyt signal occurs in platelets whose [Ca2+]mito has increased above the threshold for mPTP opening and leads to these platelets rapidly becoming procoagulant. In platelets whose [Ca2+]mito is below this threshold, mPTP remains closed, and the platelet remains noncoagulant. Graded variation in the initial [Ca2+]cyt signal between platelets is therefore converted into an all-or-nothing signal and a commitment to either becoming procoagulant or to remaining noncoagulant.
The low affinity for Ca2+ of Fluo-5N is necessary to identify the supramaximal [Ca2+]cyt signal in procoagulant platelets. Using high-affinity Fluo-4, we could not detect a distinct difference in apparent [Ca2+]cyt between procoagulant and noncoagulant platelets. Although the median Fluo-4 fluorescence of procoagulant platelets was often higher than the median fluorescence of noncoagulant platelets, there was considerable overlap in their fluorescence distributions. Based on Fluo-4, we might conclude that although high [Ca2+] is required to generate procoagulant platelets, there must be other signals that commit a platelet to becoming procoagulant. However, when we used the low-affinity Ca2+ dye, Fluo-5N, a clear difference in fluorescence was noted between procoagulant and noncoagulant platelets. Based on the in vitro Kd of Fluo-5N, we estimate the [Ca2+]cyt signal in procoagulant platelets as being >100 µM. This value is much larger than anything previously reported in platelets and much higher than 1 to 2 µM, the apparent maximal [Ca2+]cyt often reported by using high-affinity dyes. It is also >250× the Kd of Fluo-4, far beyond the range that Ca2+-sensitive dyes are likely to be reliable.21 Fluo-4 would be saturated before supramaximal [Ca2+]cyt levels are reached. The same difficulty applies to Fura-red (used by Obydennyi et al9 ), which has a similar high affinity to Fluo-4.
One implication of such a high [Ca2+]cyt signal in procoagulant platelets is that it may activate effectors with relatively low Ca2+ affinity. In particular, supramaximal [Ca2+]cyt signaling may be necessary to expose PS because the phospholipid scramblase is poorly Ca2+ sensitive. The half-maximal Ca2+ sensitivity of phospholipid scrambling, or of TMEM16F, the scramblase in platelets, red blood cells, and lymphocytes26,27 has been estimated between 10 and 80 µM.28-31 With this low Ca2+ sensitivity, it is unclear how it might be effectively activated by the low micromolar [Ca2+]cyt previously suggested by high-affinity Ca2+ dyes. However, the scramblase could be effectively activated by the concentrations reached during supramaximal [Ca2+]cyt signaling.
Supramaximal Ca2+ signaling has been observed in other cells. [Ca2+]cyt >100 µM was reported in adenosine triphosphate–depleted renal proximal tubular cells32 and MDCK cells33 by using low-affinity dyes. In excitotoxic neurons, [Ca2+]cyt was estimated as >5 µM by using a low-affinity dye but only 0.3 to 0.4 µM with high-affinity fura-2; cell death correlated with [Ca2+]cyt reported by using the low-affinity dye.34 A consistent feature of these reports is that the cells were undergoing cell death, and procoagulant platelets are described as necrotic.35,36 Importantly, however, supramaximal [Ca2+]cyt signaling is not simply a consequence of necrotic platelets losing plasma membrane integrity, as shown by their retention of calcein. Consistent with this finding, although procoagulant platelets stain with the cell-impermeable cell death marker 4-[N-(S-glutathionylacetyl)amino] phenylarsonous acid, entry of this cell death marker required organic anion transporters,35 also indicating that the plasma membrane remains intact during the early stages of procoagulant platelet formation.
Supramaximal [Ca2+]cyt signaling requires mPTP opening, meaning that it only occurs in those platelets in which [Ca2+]mito has exceeded the threshold. We could readily repeat the observation that the mPTP opening is required for procoagulant platelets, as CsA reduced the percentage of platelets that became procoagulant, consistent with previous reports with CsA and with CypD-deficient mouse platelets.5,6,35 In our study, CsA also reduced the percentage of platelets with high Fluo-5N fluorescence. This finding indicates that mPTP opening leads to supramaximal [Ca2+]cyt signaling. In contrast, if supramaximal [Ca2+]cyt signaling triggered mPTP opening, it would be expected to have no effect on the total percentage of platelets with high Fluo-5N fluorescence.
Although CsA reduced the percentage of platelets that became procoagulant, the inhibition was not complete. This is likely because CsA inhibits CypD, which normally reduces the threshold of [Ca2+]mito required for mPTP opening. When platelets are treated with CsA, the threshold of [Ca2+]mito required for mPTP opening will increase. Although this prevents most platelets from becoming procoagulant, a small percentage may have a sufficiently high initial [Ca2+]cyt signal that the [Ca2+]mito increases beyond the higher threshold. In these few platelets, mPTP opens, and a supramaximal [Ca2+]cyt signal is generated. In these platelets, the Fluo-5N fluorescence is similar to that in procoagulant platelets without CsA, or slightly higher.
Platelet Ca2+ signaling is often measured in cuvettes or microplates, in which the fluorescence is the combined signal from all the platelets, both procoagulant and noncoagulant. The inhibitory effect of CsA can be readily seen by this approach when Fluo-5N was used. In contrast, CsA did not seem to inhibit Ca2+ signaling when Fluo-4 was used. This is because there was considerable overlap in the Fluo-4 fluorescence distribution of procoagulant and noncoagulant platelets. These Fluo-4 data are consistent with those of Jobe et al,5 who reported that [Ca2+]cyt signaling was not obviously different in CypD-deficient mouse platelets when using Fluo-4 as the Ca2+ indicator. However, we suggest that [Ca2+]cyt is, in fact, much higher in procoagulant platelets, which can be shown with a low-affinity Ca2+ indicator.
Our model, and the observation that experiments with high-affinity indicators in cell populations do not readily report the effect of inhibition of supramaximal [Ca2+]cyt signaling, prompted us to reassess the contribution of 2 different Ca2+ channels to platelet procoagulant signaling. Synta-66 was used to assess the role of Orai1, which is responsible for store-operated Ca2+ entry (SOCE) in platelets.24,37,38 Consistent with previous reports, inhibition of SOCE reduced the proportion of platelets that become procoagulant and inhibited the total fluorescence in Fluo-4–loaded platelets.22,25,37 Synta-66 also slightly inhibited the Fluo-4 fluorescence in noncoagulant platelets. It had no effect on the supramaximal [Ca2+]cyt seen in those platelets that did become procoagulant. This finding suggests that SOCE through Orai1 contributes to the initial [Ca2+]cyt signal. Inhibition of SOCE leads to reduced [Ca2+]mito, fewer platelets exceeding the mPTP threshold, fewer platelets generating a supramaximal [Ca2+]cyt signal, and therefore fewer platelets becoming procoagulant. Those platelets in which mPTP opens have a normal supramaximal [Ca2+]cyt signal. This further suggests that SOCE does not play a direct role in generating the supramaximal [Ca2+]cyt signal. SOCE is not a suitable target to selectively inhibit platelet procoagulant activity because it is involved in the initial [Ca2+]cyt signal.
The MCU is required for Ca2+ uptake into mitochondria. Consistent with previous reports,5,13 the MCU inhibitor Ru360 reduced the percentage of platelets that became procoagulant. However, unlike Synta-66, it had no effect on the Fluo-4 fluorescence in the total platelet population, indicating that MCU is not involved in the initial [Ca2+]cyt signal; rather, by reducing mitochondrial Ca2+ uptake, it reduces the proportion of platelets whose [Ca2+]mito exceeds the mPTP threshold. Those platelets in which mPTP opens have a normal supramaximal [Ca2+]cyt signal. This further suggests that MCU does not play a direct role in generating the supramaximal [Ca2+]cyt signal. However, because MCU is also not involved in the initial [Ca2+]cyt signal, it is a potential target to selectively reduce platelet procoagulant activity.
Many other signaling pathways have been proposed to regulate platelet procoagulant activity to some extent, and these may also be reinterpreted in terms of this model. These signaling pathways include other plasma membrane channels, such as TRPC6,23,38 Cl– channels,20 and aquaporins39 ; plasma membrane receptors, including protease-activated receptor 1,40,41 protease-activated receptor 4,42,43 P2Y12,44 and integrin αIIbβ345-47 ; signaling platforms such as lipid rafts18,48 and diverse intracellular signaling proteins such as Rac149 ; different protein kinase C isoforms7,50,51 ; and nuclear receptors.52 We suggest that although many of these pathways are likely to regulate the initial [Ca2+]cyt signal, it may be useful to reassess the contribution of these pathways because they may also reveal targets to selectively inhibit platelet procoagulant activity.
In conclusion, our findings illustrate supramaximal Ca2+ signaling in procoagulant platelets. Although it is not yet clear how mPTP opening leads to the supramaximal Ca2+ signal (and it may be release of Ca2+ from mitochondria themselves53 ), our model provides a new framework to interpret the role of different platelet signaling systems in procoagulant platelet formation.
Acknowledgments
This work was supported by the British Heart Foundation (PG/17/45/33071 [N.A.], FS/15/62/32032 [S.L.M.-B.], and SP/15/7/31561 [J.-D.M.]).
Authorship
Contribution: N.A. designed and performed experiments, analyzed data, and wrote and edited the manuscript; S.L.M.-B. designed and performed experiments, analyzed data, and edited the manuscript; S.C. performed experiments and analyzed data; J.-D.M. synthesized CRP-XL; and M.T.H. designed experiments and wrote and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Matthew T. Harper, Department of Pharmacology, Tennis Court Rd, Cambridge CB2 1PD, United Kingdom; e-mail: mth29@cam.ac.uk.

