

Key Points
Skeletal muscle myosin binds von Willebrand factor.
Skeletal muscle myosin does not directly bind factor VIII.

Abstract
von Willebrand factor (VWF) binds to platelets and collagen as a means of facilitating coagulation at sites of injury. Recent evidence has shown that myosin can serve as a surface for thrombin generation and binds to activated factor V and factor X. We studied whether VWF can also bind myosin as a means of bringing factor VIII (FVIII) to sites of clot formation. A myosin-binding assay was developed using skeletal muscle myosin to measure VWF binding, and plasma-derived and recombinant VWF containing molecular disruptions at key VWF sites were tested. Competition assays were performed using anti-VWF antibodies. FVIII binding to myosin was measured using a chromogenic FVIII substrate. Thrombin generation was measured using a fluorogenic substrate with and without myosin. Wild-type recombinant VWF and human plasma VWF from healthy controls bound myosin, whereas plasma lacking VWF exhibited no detectable myosin binding. Binding was multimer dependent and blocked by anti-VWF A1 domain antibodies or A1 domain VWF variants. The specific residues involved in myosin binding were similar, but not identical, to those required for collagen IV binding. FVIII did not bind myosin directly, but FVIII activity was detected when VWF and FVIII were bound to myosin. Myosin enhanced thrombin generation in platelet-poor plasma, although no difference was detected with the addition of myosin to platelet-rich plasma. Myosin may help to facilitate delivery of FVIII to sites of injury and indirectly accelerate thrombin generation by providing a surface for VWF binding in the setting of trauma and myosin exposure.
Introduction
von Willebrand factor (VWF) has known binding sites for 3 main ligands. VWF binds to factor VIII (FVIII) and protects FVIII from degradation in plasma through a binding site in the VWF D′D3 domain.1 VWF binds platelet glycoprotein Ibα and, thus, enables delivery of platelets to sites of injury through a binding site in the VWF A1 domain.2 VWF also binds to subendothelial collagen, which is exposed at sites of injury, through binding sites for types I and III collagen in the VWF A3 domain3 and types IV and VI collagen in the VWF A1 domain.4 von Willebrand disease (VWD) can result from a defect in any of these functions.5 Platelet binding is measured most commonly by the VWF ristocetin cofactor activity assay, but newer methods using gain-of-function GPIb have gained traction as a result of their greater reliability.5,6 Defects in platelet binding are arguably the most common VWF defect and are the most critical to measure as a diagnostic test; however, other functional defects can cause VWD. Defects in VWF–collagen interactions have been demonstrated for several vascular collagens (I, III, IV, and VI).7,8
Recent evidence has shown that myosin can serve as a surface for thrombin generation.9 Work by Griffin and colleagues demonstrated a role for myosin in binding activated FV and activated FX.9 This mimics the role of collagen IV in binding FIX, as demonstrated by Stafford and colleagues.10,11 Griffin and colleagues also demonstrated increased myosin-related thrombin generation in the setting of trauma,9 a situation in which muscle myosin might be exposed. This suggests that trauma affecting muscles will expose myosin, which could subsequently affect hemostasis. In this study, we examined whether VWF could also bind myosin as a means of delivering FVIII to sites of clot formation. If true, this could mean that myosin serves as a surface, much like the platelet phospholipid membrane, and could catalyze the ability of VWF to bring FVIII to sites of clot formation in a manner similar to VWF–collagen interactions.
Methods
Generation of recombinant variant VWF
VWF variants containing specific point mutations known to alter VWF function were constructed via site-directed mutagenesis, as previously described.12 Recombinant wild-type (WT) VWF and variant VWF constructs were transfected into HEK293T cells, and supernatants were collected for VWF to use in experiments. Table 1 denotes the variants studied, their location, and their typical effect on VWF function. The variant p.Y87S lacks the ability to form C-terminal dimers.13 The p.C2773R variant was created to disrupt C-terminal dimerization, but it can also form dimers through the N-terminal intact binding site.
VWF variants used in myosin-binding experiments
| Variant | Location in VWF structure | Typical effect on VWF function |
|---|---|---|
| p.Y87S | D1 domain | Impairs N-terminal multimerization |
| p.T791M | D′ domain | Removes ability of VWF to bind FVIII |
| p.V1316M | A1 domain | Type 2B variant, increases VWF binding to platelet GPIb |
| p.F1369I | A1 domain | Type 2M variant, decreases VWF binding to platelet GPIb |
| p.R1392A | A1 domain | Removes ability of VWF to bind type IV collagen |
| p.R1395A | A1 domain | Removes ability of VWF to bind type IV collagen |
| p.R1399A | A1 domain | Removes ability of VWF to bind type IV collagen |
| p.R1399H | A1 domain | Removes ability of VWF to bind type IV collagen |
| p.R1402A | A1 domain | Removes ability of VWF to bind type IV collagen |
| p.H1786D | A3 domain | Removes ability of VWF to bind type III collagen |
| p.D2509E | C domain (C4) | Alters RGD site, removes binding to platelet αIIbβ3 |
| p.C2773R | C domain | Impairs C-terminal dimerization |
| Variant | Location in VWF structure | Typical effect on VWF function |
|---|---|---|
| p.Y87S | D1 domain | Impairs N-terminal multimerization |
| p.T791M | D′ domain | Removes ability of VWF to bind FVIII |
| p.V1316M | A1 domain | Type 2B variant, increases VWF binding to platelet GPIb |
| p.F1369I | A1 domain | Type 2M variant, decreases VWF binding to platelet GPIb |
| p.R1392A | A1 domain | Removes ability of VWF to bind type IV collagen |
| p.R1395A | A1 domain | Removes ability of VWF to bind type IV collagen |
| p.R1399A | A1 domain | Removes ability of VWF to bind type IV collagen |
| p.R1399H | A1 domain | Removes ability of VWF to bind type IV collagen |
| p.R1402A | A1 domain | Removes ability of VWF to bind type IV collagen |
| p.H1786D | A3 domain | Removes ability of VWF to bind type III collagen |
| p.D2509E | C domain (C4) | Alters RGD site, removes binding to platelet αIIbβ3 |
| p.C2773R | C domain | Impairs C-terminal dimerization |
Myosin enzyme-linked immunosorbent assay
Myosin (rabbit skeletal muscle, myosin II; Cytoskeleton, Denver, CO) was plated on maleic anhydride plates and allowed to incubate at 4°C overnight. Plates were washed and blocked with bovine serum albumin. Plasma or recombinant VWF was added and incubated at room temperature for 1 hour. The presence of bound VWF was detected using biotinylated anti-VWF monoclonal antibodies (AVW1 and AVW15; Blood Research Institute). VWF plasmas tested included healthy control donor plasmas, as well as plasma from donors with type 1 and type 2A VWD. Recombinant VWF preparations tested included WT and various VWF variants.
Competition assays were performed using antibodies known to block various sites on VWF; VWF and the antibody were incubated together for 20 minutes prior to their addition to the enzyme-linked immunosorbent assay (ELISA) plate. Antibodies used included AVW3, AVW5, AP2 (Blood Research Institute), and polyclonal anti-VWF (Dako, Carpinteria, CA). Additional testing was performed using platelet-poor plasma from healthy controls and individuals with type 3 VWD.
Collagen inhibition
Myosin ELISA was performed as above, with the addition of collagen IV or collagen III (both from SouthernBiotech, Birmingham, AL). Individual collagen preparations were added at a concentration of 25 mcg/mL at the same time as the VWF solution and incubated at room temperature for 1 hour.
FVIII activity
Plates were coated with myosin as described above. Two different recombinant FVIII preparations (preparation 1, Kogenate; Bayer, Whippany, NJ and preparation 2, XYNTHA; Pfizer, New York, NY) were tested, as well as a plasma-derived purified concentrate containing VWF and FVIII (HUMATE-P; CSL Behring, King of Prussia, PA). Bound FVIII coagulant activity was measured using a chromogenic substrate (Chromogenix Coatest; Diapharma, West Chester, OH).14
Thrombin generation
Thrombin generation was assessed using a Technothrombin TGA Kit (Diapharma).15 A fluorogenic substrate was added to platelet-poor plasma in the presence of tissue factor alone (recombinant human tissue factor; Haemtech Biopharma, Essex Junction, VT) or tissue factor plus myosin (200 nM). Additional thrombin-generation studies were done using replication-competent lentivirus (RCL) reagent from the Technothrombin Kit (Diapharma), which contains tissue factor and phospholipids, with the latter at a lower concentration compared with their other reagents.
Binding affinity
VWF binding to myosin was measured using Octet Red 96 (ForteBio, San Jose, CA). Myosin (10 mcg/mL) was bound to an amine reactive sensor (ARG2; ForteBio) using their Amine Coupling Kit and pH 4 buffer. Recombinant WT VWF was diluted to concentrations ranging from 0.055 mcg/mL to 3.5 mcg/mL. A 1:1 binding model was used according to ForteBio Data Analysis software (v9.0).16
Statistics
Statistical analyses were performed using GraphPad Prism 8.0.0 for Windows (GraphPad Software, San Diego, CA). The Student t test was used to compare data sets for the thrombin-generation assay. Pairwise comparisons were performed using a Tukey SD post hoc test (SAS, Cary, NC) to test the mean differences in pairwise groups for the VWF constructs.
Results
VWF binds to myosin
When skeletal muscle myosin was bound to a plate, VWF in plasma from healthy individuals was detected, with a ratio of myosin binding to VWF antigen (VWF:Ag) ∼1 (Figure 1A). Results are presented as a ratio to account for varying amounts of VWF protein (VWF:Ag) found in different plasmas and produced by different constructs. No VWF binding was seen with plasma from individuals with type 3 VWD, which was included as a negative control. Plasma from individuals with type 1 VWD showed a ratio similar to that seen in healthy controls, although the former had VWF:Ag < 30 IU/dL. In contrast, plasma from individuals with type 2A VWD showed a decreased myosin binding to VWF:Ag ratio, similar to ratios seen with platelet or collagen binding in these patients.
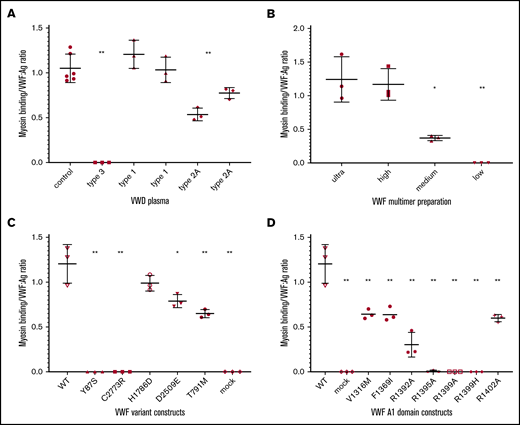
Myosin binds to VWF. VWF:Ag is shown for plasma-derived VWF and recombinant VWF. (A) Myosin binding in plasma samples from healthy donors (“control”) and donors with VWF (types 3, 1, and 2A). No significant difference was found between the control myosin binding and type 1 myosin binding, but control myosin binding and type 1 myosin binding were significantly different compared with type 2 myosin binding. (B) Myosin binding is dependent on presence of high molecular weight multimers using recombinant VWF separated into fractions based on the molecular weight of the multimers contained therein (ultra high, high, medium, and low). A significant difference was noted for the medium and low molecular weight multimers compared with the high and ultrahigh molecular weight multimers. (C) Myosin binding to recombinant VWF, including WT recombinant VWF, constructs containing VWF variants that affect multimerization (87S and 2773R), the VWF A3 domain (1786D), which affects collagen III binding, a C-terminal domain RGD site variant (2509E), a D′ variant causing type 2N VWD (791M), and a construct without any VWF sequence as a negative control (mock). (D) A1 domain specificity of myosin binding to recombinant VWF constructs. n ≥ 3. Error bars denote 1 standard deviation. *P < .05, **P < .005.
Myosin binds to VWF. VWF:Ag is shown for plasma-derived VWF and recombinant VWF. (A) Myosin binding in plasma samples from healthy donors (“control”) and donors with VWF (types 3, 1, and 2A). No significant difference was found between the control myosin binding and type 1 myosin binding, but control myosin binding and type 1 myosin binding were significantly different compared with type 2 myosin binding. (B) Myosin binding is dependent on presence of high molecular weight multimers using recombinant VWF separated into fractions based on the molecular weight of the multimers contained therein (ultra high, high, medium, and low). A significant difference was noted for the medium and low molecular weight multimers compared with the high and ultrahigh molecular weight multimers. (C) Myosin binding to recombinant VWF, including WT recombinant VWF, constructs containing VWF variants that affect multimerization (87S and 2773R), the VWF A3 domain (1786D), which affects collagen III binding, a C-terminal domain RGD site variant (2509E), a D′ variant causing type 2N VWD (791M), and a construct without any VWF sequence as a negative control (mock). (D) A1 domain specificity of myosin binding to recombinant VWF constructs. n ≥ 3. Error bars denote 1 standard deviation. *P < .05, **P < .005.
Given the decreased myosin binding in plasma missing high molecular weight multimers, multimer dependence was investigated further (Figure 1B) using purified VWF multimer fractions, with solutions primarily containing ultra-high molecular weight multimers, high molecular weight multimers, medium molecular weight multimers, and low molecular weight multimers. The binding appeared to be dependent on multimer size, because solutions with varying multimer distributions demonstrated a size-dependent decrease in VWF binding. The highest binding was seen with a solution of ultra-high molecular weight multimers, whereas binding was undetectable with a solution of only low molecular weight multimers (Figure 1B).
The region of VWF responsible for myosin binding was probed using VWF variants with specific defects (Figure 1C). As expected given the multimer results, 2 VWF variants that affect N-terminal multimerization and C-terminal dimerization, p.87S and p.2773R, did not bind to myosin. VWF variant p.H1786D, which is located in the VWF A3 domain, showed normal binding to myosin, although this variant does not bind collagen III. A statistically significant decrease in binding was seen with p.D2509E, which eliminates binding to platelet αIIbβ3. Decreased binding was also seen with p.T791M, a variant in the D′ domain that affects binding to FVIII.
VWF A1 domain variants are shown separately in Figure 1D. Decreased or absent binding was seen with several VWF A1 domain variants. A significant decrease in binding was seen with p.V1316M and p.F1369I, which are variants seen in type 2B and type 2M VWD. We then investigated a series of A1 domain variants that abrogate VWF binding to collagen IV. VWF variants p.R1395A and p.R1399A, as well as the relatively common variant p.R1399H, did not have detectable binding to myosin. A significant reduction in binding was seen with variants p.R1392A and p.R1402A (Figure 1D); however, there was still some residual binding, in contrast to their effect on collagen IV binding.
VWF binding to myosin was blocked by anti-VWF antibodies, with inhibition of binding seen using a polyclonal antibody (DAKO), as well as an antibody directed against the VWF A1 domain (AVW3) (Figure 2). Antibodies directed against other VWF sites, including the N- and C-terminal ends, failed to affect VWF binding to myosin. Addition of collagen IV completely blocked VWF binding to myosin, as expected given the similarity in the binding site in the VWF A1 domain. Unexpectedly, collagen III also inhibited VWF binding to myosin (Figure 2).
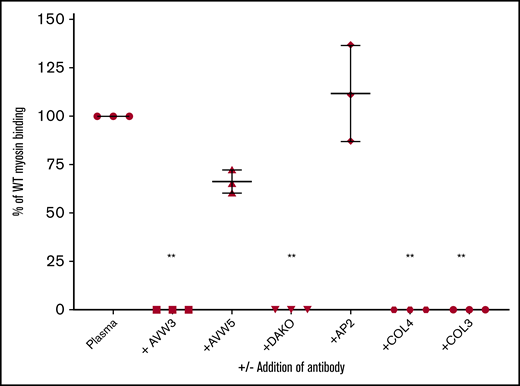
VWF binding to myosin inhibited by anti-A1 domain antibodies and collagen. VWF binding to myosin was measured with or without the addition of anti-VWF antibodies or collagen. The y-axis shows the percentage of VWF bound to myosin compared with a sample with VWF and no inhibitor. AVW3 is an anti-VWF A1 domain antibody. AVW5 is also an anti-VWF antibody, but it is located outside of the A1 domain. DAKO is a polyclonal anti-VWF antibody. AP2 is an anti-platelet αIIbβ3 antibody added as a negative control. Collagen IV (COL4) and collagen III (COL3) were also added as potential inhibitors. Error bars denote 1 standard deviation. n ≥ 3 for each experiment. **P < .005 vs control plasma (Plasma).
VWF binding to myosin inhibited by anti-A1 domain antibodies and collagen. VWF binding to myosin was measured with or without the addition of anti-VWF antibodies or collagen. The y-axis shows the percentage of VWF bound to myosin compared with a sample with VWF and no inhibitor. AVW3 is an anti-VWF A1 domain antibody. AVW5 is also an anti-VWF antibody, but it is located outside of the A1 domain. DAKO is a polyclonal anti-VWF antibody. AP2 is an anti-platelet αIIbβ3 antibody added as a negative control. Collagen IV (COL4) and collagen III (COL3) were also added as potential inhibitors. Error bars denote 1 standard deviation. n ≥ 3 for each experiment. **P < .005 vs control plasma (Plasma).
FVIII binding is not affected by VWF–myosin interactions
FVIII activity was detected when a VWF concentrate containing VWF and FVIII was bound to myosin, with a dose-dependent increase in activity (Figure 3). No FVIII activity was detected when FVIII was added to myosin in the absence of VWF. These results suggest that the increase in FVIII activity is due to VWF delivering its endogenously bound FVIII to the immobilized myosin, not to an intrinsic relationship between myosin and FVIII.
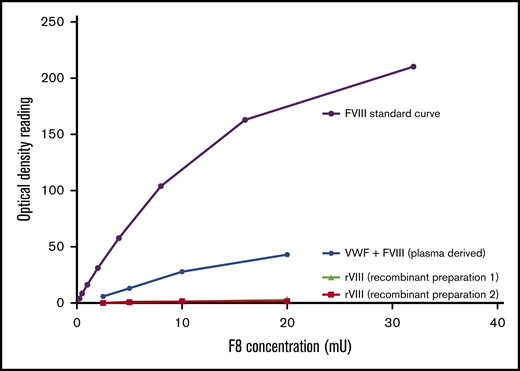
FVIII binding to myosin detected only in presence of VWF. FVIII activity was measured with a chromogenic FVIII activity kit. Recombinant FVIII or plasma-derived combination VWF/FVIII concentrate was added to a plate coated with myosin. Bound FVIII was detected using a chromogenic FVIII activity kit. No activity was seen when only FVIII was added (preparation 1, full-length FVIII; preparation 2, B-domain–deleted FVIII), but FVIII activity was detected in the presence of VWF.
FVIII binding to myosin detected only in presence of VWF. FVIII activity was measured with a chromogenic FVIII activity kit. Recombinant FVIII or plasma-derived combination VWF/FVIII concentrate was added to a plate coated with myosin. Bound FVIII was detected using a chromogenic FVIII activity kit. No activity was seen when only FVIII was added (preparation 1, full-length FVIII; preparation 2, B-domain–deleted FVIII), but FVIII activity was detected in the presence of VWF.
Thrombin generation is not affected by VWF–myosin interactions
Because previous results from Griffin and coworkers showed a role for myosin in enhancing thrombin generation,9 we also assessed thrombin generation with and without myosin. We did not see an increase in thrombin generation in platelet-rich plasma with the use of tissue factor for initiation. However, we did note increased thrombin generation in the setting of platelet-poor plasma, with an average total thrombin generation (area under the curve) of 613 for platelet-poor plasma without added myosin and 1053 with the addition of myosin, a nearly twofold increase (Figure 4). When the RCL reagent from the Thrombin Generation Kit containing tissue factor and phospholipids was used, there was again no difference between plasma with added myosin and plasma without added myosin (data not shown). The main difference appears to be in the setting of platelet-poor plasma, in which the addition of myosin results in significantly greater thrombin generation.
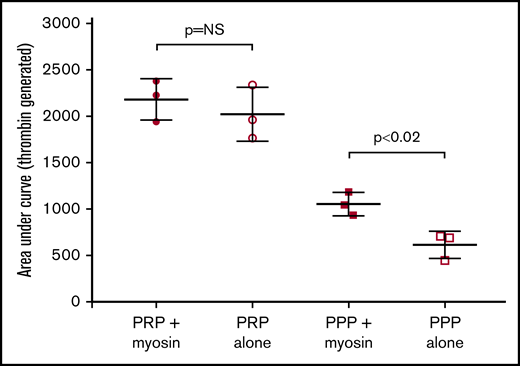
Myosin enhances thrombin generation in the absence of platelets. Thrombin generation was performed for platelet-rich plasma (PRP) and platelet-poor plasma (PPP), with or without the addition of 200 nM myosin. All samples had tissue factor added to initiate thrombin generation. The graph shows total thrombin generated (area under the curve). No difference was seen for PRP, but addition of myosin increased thrombin generation for PPP. Error bars denote 1 SD. N ≥ 3 for each experiment. NS, not significant.
Myosin enhances thrombin generation in the absence of platelets. Thrombin generation was performed for platelet-rich plasma (PRP) and platelet-poor plasma (PPP), with or without the addition of 200 nM myosin. All samples had tissue factor added to initiate thrombin generation. The graph shows total thrombin generated (area under the curve). No difference was seen for PRP, but addition of myosin increased thrombin generation for PPP. Error bars denote 1 SD. N ≥ 3 for each experiment. NS, not significant.
The binding affinity of myosin for VWF was ascertained using the Octet biosensor (Table 2). The average KD for VWF bound to myosin was 7.0 × 10−9 M.
Discussion
Our results demonstrate that myosin is able to bind VWF and may have a physiologic role in clot formation during vascular injury. Myosin–VWF interactions may to help facilitate delivery of FVIII to sites of injury. When trauma occurs, myosin may be exposed, as supported by data from Griffin and colleagues showing increased thrombin-generation potential due to myosin in trauma patients.9 Our data show that myosin binds VWF via the VWF A1 domain in a slightly different location than collagen IV and VI.8,17 This may be secondary to steric hindrance or, it may, in fact, represent binding to different sets of A1 domain amino acids in the VWF protein; however, it does suggest that the binding sites for myosin and collagen IV on VWF are not identical.17 Residues that affect myosin binding are not necessarily those that affect binding to platelet GPIb, although platelet GPIb also binds VWF via the A1 domain.18 Numerous residues have been implicated in reducing VWF interactions with platelet GPIb,19 whereas, at least with human samples containing the p.R1399H variant, platelet binding is normal.17 Interestingly, the KD of VWF bound to myosin (7 nM) was similar to that previously reported for VWF A1 domain binding to collagen type I (8 nM).20
Myosin binding to VWF was also dependent on multimer size. This is not surprising, given the requirement for high molecular multimers to achieve optimal platelet and collagen binding.21 The improved activity of high molecular weight multimer forms of VWF in platelet binding activity, in particular, is important, given the bleeding seen in type 2A VWD patients who lack high molecular weight multimers.22 Our data also suggest that dimer forms of VWF, and likely monomeric VWF, do not bind myosin. Conceptually, higher molecular weight multimers would also unfold to a longer length, providing a greater ability to recruit platelets to sites of injury.23
Antibodies against the VWF A1 domain blocked VWF binding to myosin. In addition, VWF binding to myosin was abrogated by sequence variants in the VWF A1 domain at 1395 and 1399 amino acids, similar to the effect of these variants on collagen IV binding.17 However, other residues that reduced or removed collagen IV binding (p.1392A and p.1402A) had a lesser effect on myosin binding. Taken together, these results suggest a binding site for myosin in the VWF A1 domain that is similar, but not identical, to that of collagen IV. Although collagen III is classically known to bind the VWF A3 domain, there is some evidence that it possesses a binding site in the VWF A1 domain.24 However, because collagens are large molecules, it is also possible that the inhibition is due to steric hindrance. The decreased binding with the p.791M variant is interesting because this variant affects FVIII binding but is located in the VWF TIL′ domain.25
Although FVIII activity was enhanced by the combination of VWF and myosin, myosin alone did not provide a surface for FVIII activity. This supports the theory that FVIII does not bind directly but instead requires VWF to transport it to sites of clot formation; however, this mechanism contrasts with FV and FX, which bind directly to myosin.9 It also contrasts with FIX, which has been shown to bind directly to collagen IV.10 FVIII, unlike VWF, requires a membrane surface on which to function in concert with activated FIX,26 which may explain its lack of myosin binding. Alternately, one could hypothesize that because FVIII’s coproteins VWF and FIX can bind myosin and collagen, there is no need for such a binding site on FVIII.
Thrombin generation in platelet-poor plasma, when initiated by tissue factor, was enhanced by the addition of skeletal myosin. In contrast, we did not find an increase in thrombin generation in settings in which additional phospholipids were present, such as in platelet-rich plasma or when using an initiation reagent that also contains phospholipids. These results suggest that myosin’s ability to enhance thrombin generation occurs primarily in situations in which platelets are not present or when minimal platelet accumulation has occurred. The exposure of myosin is most likely an immediate reaction, because data show that, upon injury, the endothelial plasma membrane is lost.27 Different injury types could potentially result in different rates of myosin exposure; however, any muscle injury would be expected to result in myosin exposure.
VWF–myosin interactions likely do not directly impact thrombin generation. The increase in thrombin generation is much more likely to occur via the previously described interactions with activated FV and FX9 ; however, these results are limited, because myosin–VWF interactions were only studied under static conditions. Further work is required to investigate this interaction directly in the setting of muscle injury, as well as the impact of myosin on in vivo clot formation. Intriguing results from the trauma literature suggest that trauma-induced coagulopathy leads to procoagulant effects measured by thromboelastography.28 These results suggest that myosin, similarly to collagen, can increase local accumulation of VWF–FVIII and, thus, indirectly accelerate thrombin generation and clot formation.
In this light, myosin could be considered an emergency platelet, binding FV and FX and recruiting FVIII via VWF until platelet recruitment and fibrin generation via conventional means take over. To that end, the addition of myosin to promote coagulation, a situation that may already be occurring in trauma patients, could potentially prove easier than supplementation with platelets.9,28 However, in clinical practice, this may end up a being delicate balance between improving hemostasis and increasing thrombosis. Regardless, our data on VWF and myosin interactions support an increased role for myosin beyond muscle fibers.
Acknowledgments
The authors thank John Griffin and the Griffin laboratory for helpful conversations.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute (HL081588 and HL126810), the Midwest Athletes Against Childhood Cancer (MACC) Fund, and the BloodCenter Research Fund.
Authorship
Contribution: V.H.F. designed the research and wrote the manuscript; T.L.S., D.K., H.K.L., and S.F. performed experiments and edited the final manuscript; L.Z. and P.S. performed statistical analyses; and R.R.M. designed the research and edited the final version of the manuscript.
Conflict-of-interest disclosure: R.R.M. has served as a consultant for Shire. The remaining authors declare no competing financial interests.
Correspondence: Veronica H. Flood, Comprehensive Center for Bleeding Disorders, 8701 Watertown Plank Rd, PO Box 2178, Milwaukee, WI 53201-2178; e-mail: vflood@mcw.edu.

