

Key Points
Novel BET inhibitors PLX51107 and PLX2853 induce apoptosis via induction of the proapoptotic BH3-only protein BIM.
PLX51107 and PLX2853 are synergistic with ABT199/venetoclax in killing MYC-driven B-cell lymphoma cells with high BCL-2 expression.

Abstract
Although the MYC oncogenic network represents an attractive therapeutic target for lymphoma, MYC inhibitors have been difficult to develop. Alternatively, inhibitors of epigenetic/ transcriptional regulators, particularly the bromodomain and extraterminal (BET) family, have been used to modulate MYC. However, current benzodiazepine-derivative BET inhibitors (BETi) elicit disappointing responses and dose-limiting toxicity in relapsed/refractory lymphoma, potentially because of enrichment of high-risk molecular features and chemical backbone-associated toxicities. Consequently, novel nonbenzodiazepine BETi and improved mechanistic understanding are required. Here we characterize the responses of aggressive MYC-driven lymphomas to 2 nonbenzodiazepine BETi: PLX51107 and PLX2853. Both invoked BIM-dependent apoptosis and in vivo therapy, associated with miR-17∼92 repression, in murine Eµ-myc lymphomas, with PLX2853 exhibiting enhanced potency. Accordingly, exogenous BCL-2 expression abrogated these effects. Because high BCL-2 expression is common in diffuse large B-cell lymphoma (DLBCL), BETi were ineffective in driving apoptosis and in vivo therapy of DLBCL cell lines, mirroring clinical results. However, BETi-mediated BIM upregulation and miR-17∼92 repression remained intact. Consequently, coadministration of BETi and ABT199/venetoclax restored cell death and in vivo therapy. Collectively, these data identify BIM-dependent apoptosis as a critical mechanism of action for this class of BETi that, via coadministration of BH3 mimetics, can deliver effective tumor control in DLBCL.
Introduction
Despite the effective rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone (R-CHOP) treatment regimen, relapsed/refractory (R/R) disease is common in diffuse large B-cell lymphoma (DLBCL), with further intervention currently associated with poor tolerability and survival.1 Consequently, additional treatment options are required.
Cell of origin (COO) classifiers identify 3 DLBCL subsets: activated B cell (ABC), germinal center B cell (GCB), and unclassified.1 There is heterogeneity of prognosis within these, according to features associated with R-CHOP resistance, centered on MYC and BCL-2.2 High coexpression without underlying chromosomal rearrangement defines the double expressor lymphoma (DEL) subgroup, most common within ABC-DLBCL, whereas, the largely GCB-DLBCL–associated double-hit lymphoma (DHL) represent high-grade lymphomas with MYC and BCL-2 or BCL-6 translocations.2 Both DHL and DEL exhibit poor prognosis3 and are over-represented in R/R cases (∼50% DEL and 13% DHL).4
MYC, a master regulator and global amplifier of transcription,5 is dysregulated in ∼70% of cancers and functions as a critical oncogene required for sustained proliferation and lymphomagenesis.5 Methods to impair MYC have been the focus of intensive research.5 Increasingly, modulation of transcription/epigenetic regulators has been investigated, including inhibition of the bromo and extraterminal domain (BET) family.6
Following bromodomain-mediated recruitment to active chromatin (by binding acetylated lysine residues of histones and other proteins),7 the BET family (BRD2, BRD3, BRD4, and BRDt) facilitate transcription via recruitment and activation of key transcriptional/epigenetic modifiers.8 In particular, BRD4 effects regulation of super enhancer (SE)–proximal genes,9,10 including DLBCL-associated oncogenes (eg, MYC),11 and release of RNA polymerase II promoter proximal pausing via P-TEFb activation.8 Accordingly, BET inhibitors (BETi) have been shown to cause MYC downregulation and cell death, via intrinsic apoptosis, in malignant cells.6
Intrinsic apoptosis is regulated by BCL-2 family-mediated control of mitochondrial outer membrane permeabilization (MOMP), driven by the pore-forming activities of BAX, BAK, and possibly BOK.12 MOMP is suppressed by cytoplasmic sequestration and neutralization of activated BAX/BAK by prosurvival BCL-2 family members BCL-2, BCL-XL, BCL-W, MCL-1, and BFL-1/BCL-2A1.13 After apoptotic insult, BH3-only proteins (BAD, tBID, BIK, BIM, BMF, HRK, NOXA, and PUMA) de-repress BAX/BAK via selective inhibition of prosurvival BCL-2 family members and displacement of activated BAX/BAK.13 Direct activator BH3-only proteins (BIM, tBID, and PUMA) are particularly potent because of their ability to activate BAX/BAK and neutralize all prosurvival BCL-2 family members.13,14 In contrast, the remaining sensitizer BH3-only proteins demonstrate selective binding of prosurvival BCL-2 family members and invoke MOMP only when mitochondria are sufficiently primed with previously bound direct activators and/or activated BAX/BAK, via triggering their displacement.13 BETi-induced intrinsic apoptosis is associated with alleviation of miR-17∼92–mediated suppression of BIM transcription15 ; however, the role of MYC downregulation in this process is debated.15,16
Because DLBCL frequently demonstrates BCL-2 dysregulation and therapeutic resistance,3,17 BETi monotherapy is unlikely to be effective in BCL-2–high malignancies (eg, DHL/DEL or R/R disease). The benzodiazepine-derivative BETi OTX015, CPI-0610, molibresib, and mivebresib were associated with limited clinical responses and dose-limiting toxicity in R/R lymphoma.18-22 The similarity in toxicity profiles (thrombocytopenia, fatigue, and gastrointestinal disturbances)18,19 suggests that these are likely attributable to the benzodiazepine chemical backbone. To exploit the ability of BETi to modulate MYC and realize their clinical potential in DEL/DHL, novel nonbenzodiazepine BETi and strategies to overcome BCL-2–mediated treatment resistance are required.
Recently, 2 novel nonbenzodiazepine BETi, PLX51107 and PLX2853, were generated using scaffold-based, crystallography-guided design to exhibit a unique binding mode and short terminal half-life to improve tolerability.23 In biochemical assays that examined the binding of acetylated histone tails to BET proteins, PLX51107 and PLX2853 displayed potent inhibitory activity (BRD4 half maximal inhibitory concentration [IC50] = 23 nM for PLX51107 and 4.3 nM for PLX2853). PLX51107 and PLX2853 inhibited the MYC reporter activity in MV4-11 cells with an IC50 of 130 and 7.2 nM, respectively (B.P., personal email communication, March 2020). Profiling of PLX51107, a less potent precursor of PLX2853, against a lymphoma cell panel showed broad growth suppression.23 Despite these observations, detailed characterization of PLX51107s in vivo mechanism of action are currently lacking, whereas any effects of PLX2853 are yet to be reported. Here we describe their antitumor effects mediated by the BH3-only protein BIM, and synergistic activity alongside the BCL-2 inhibitor ABT199/venetoclax in BCL-2–high lymphomas.
Materials and methods
Animals
Animals were maintained in conventional barrier facilities or individually ventilated cages for immunocompromised strains. Experiments were approved by local ethical committees, reporting to the Home Office Animal Welfare Ethical Review Board, and conducted according to the National Centre for the Replacement, Refinement, and Reduction of Animals in Research and Animal Research: Reporting of In Vivo Experiments guidelines under PPLs P4D9C89E and P81E129B7. Eµ-myc lymphoma cells (2 × 106) were administered to 8- to 10-week-old female C57BL/6 mice (Charles River, Saint-Germain-Nuelles, France) via intravenous injection, with treatment starting 4 days later. Animals were euthanized after detection of splenomegaly and behavioral symptoms of disease. Human SUDHL6 or OCI-LY8 cells (1 × 107) were administered to female NOD.Cg-PrkdcscidIl2rgtm1WjI/SzJ (NSG) animals (Charles River) via subcutaneous injection. Tumor volume was calculated according to the equation volume (mm3) = (length [mm] × width [mm]2)/2. Tumor volume > 1500 mm3 defines the experimental end point. Treatment started after detection of palpable tumors. PLX51107 (10 mg/kg) and PLX2853 (5 mg/kg) were administered by oral gavage once daily. PLX51107 or PLX2853 was dissolved in n-methyl-2-pyrrolidone and subsequently added 1:9 to a diluent consisting 40% polyethylene glycol-400, 5% tocopherol methoxypolyethylene glycol succinate, and 5% Poloxamer 407. ABT199 (50 mg/kg) was administered once daily in 60% phosal-50PG, 30% polyethylene glycol-400, and 10% ethanol by oral gavage. Compounds were administered 5 consecutive d/wk as indicated in the figure legends.
Cell culture
Eμ-myc lymphoma cell lines were generated and cultured as previously described.24 BH3-only−/− Eμ-myc lymphoma cell lines were generously gifted by C. Scott and A. Strasser. SUDHL6, DOHH2, BJAB, BL60, and BL41 cells were the generous gift of G. Packham and were maintained in RPMI supplemented with 10% fetal calf serum, 2 mM l-glutamine, 1 mM sodium pyruvate, 100 U/mL penicillin, and 100 µg/mL streptomycin (Life Technologies, Paisley, United Kingdom) at 37°C, 5% CO2. OCI-LY1 and OCI-LY8 were also gifted by G. Packham and were cultured in Iscove modified Dulbecco medium supplemented with 20% human AB serum (Sigma Aldrich, Gillingham, United Kingdom), l-glutamine, sodium pyruvate, penicillin, and streptomycin at 37°C, 5% CO2. Cell line identity was confirmed using short tandem repeat (STR) analysis using the Powerplex 16 System (Promega, Southampton, United Kingdom).
Western blot and immunoprecipitation
Cells (5 × 106 for western blot or 1 × 108 for immunoprecipitation) were incubated as required, and western blotting and immunoprecipitation were undertaken using standard techniques, as previously described.25 For immunoprecipitation experiments, lysis was undertaken using 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) lysis buffer (20 mM Tris pH 7.4, 142.5 mM KCl, 2 mM CaCl2, 1% CHAPS) with anti-human BCL-2 (clone: 6C8) and anti-mouse BCL-2 (clone: 3F11) (both BD Biosciences, Oxford, United Kingdom) used as the precipitating antibody vs an anti-human CD18 isotype control (HB226). Coimmunoprecipitated proteins were detected using Veriblot horseradish peroxidase–conjugated antibodies (Abcam, Cambridge, United Kingdom).
Cellular assays and retroviral transduction
Annexin V/propidium iodide (PI) and DiOC6/ PI viability assays, hypotonic PI cell cycle assays, and BIM RNAi via retroviral transduction were performed as previously described.24-26 For further details and other methods, see the supplemental Methods.
Results
PLX51107 and PLX2853 modulate BET biomarker expression
To investigate the therapeutic potential and mechanisms of action of PLX51107 and PLX2853, aggressive MYC-driven lymphoma models were used; namely, established human B-lymphoma cell lines (supplemental Tables 1 and 2) and spontaneous IgM+ murine Eμ-myc lymphoma cell lines (LCLs) derived from transgenic (Tg) animals.27 PLX51107 exhibited a maximum plasma concentration of ∼50 μM 1 hour after treatment, falling to ∼10 μM after 8 hours after a single 20-mg/kg dose in mice.23 To assess on-target activity, validated BET biomarker (HEXIM1 and MYC) expression was assessed at physiologically relevant concentrations (≤10 µM).6,28 Both PLX51107 and PLX2853 increased HEXIM1 mRNA and decreased MYC mRNA and protein expression in human LCLs (supplemental Figure 1A-C). PLX2853 exhibited enhanced potency, with increases in HEXIM1 detectable at lower concentrations. Similar increases in HEXIM1 transcript and reduction of MYC protein expression were also observed in Eμ-myc LCLs (supplemental Figure 1D-E). These observations indicate effective BET inhibition in both human and murine lymphoma models.
PLX51107 and PLX2853 elicit intrinsic apoptosis in Eμ-myc lymphoma cell lines
Administration of benzodiazepine-derivative BETi (JQ1 and OTX015) has been associated with cell death.15,29 Consistent with these effects, PLX51107 and PLX2853 reduced viability (staining exemplified in supplemental Figure 2A) in Eμ-myc LCLs, with PLX2853 exhibiting greater potency than PLX51107 and benzodiazepine-derivative BETI (Figure 1A-B). PLX51107- and PLX2853-induced cell death displayed hallmarks of apoptosis,30 as death was abrogated by the pan-caspase inhibitor qVD (Figure 1C). To further dissect their BETi-induced apoptosis, Eμ-myc LCLs were engineered to overexpress (OE) negative regulators of intrinsic apoptosis, BCL-2, or BCL-XL (supplemental Figure 3A-B). Crucially, these engineered cell lines retained BETi-mediated increases in HEXIM1 mRNA and reduction in MYC protein levels (supplemental Figure 3C-D). Analogous to qVD, BCL-2 or BCL-XL OE abrogated PLX51107- and PLX2853-induced cytotoxicity (Figure 1D). These data indicate that PLX51107 and PLX2853 invoke cell death via intrinsic or type II extrinsic apoptosis. To scrutinize this, BETi-induced change in mitochondrial membrane potential (Δψm), indicative of MOMP, was examined (supplemental Figure 2B). PLX51107 and PLX2853 invoked substantial Δψm that was ameliorated by BCL-2 or BCL-XL OE but not by qVD (Figure 1E). Consequently, intrinsic apoptosis (requiring prior caspase-independent MOMP and downstream caspase activation) represents their mechanism of cell killing. Consistent with this, both PLX51107 and PLX2853 showed significant in vivo therapeutic responses in wild-type, but not BCL-2 OE, Eμ-myc lymphoma-bearing mice (Figure 1F). Thus, similarly to benzodiazepine-derivative BETi,15 intrinsic apoptosis was identified as the therapeutic mechanism for PLX51107 and PLX2853 in Eμ-myc lymphomas.
![PLX51107 and PLX2853 BETi invoke cell death via intrinsic apoptosis in Eμ-myc lymphomas. (A) PLX51107 (PLX5), PLX2853 (PLX2), JQ1, and OTX015 were administered to Eμ-myc lymphoma cell lines for 48 hours, and viability was assessed by annexin V/PI staining vs vehicle control (dimethyl sulfoxide [DMSO]). Points represent the mean of 12 wild-type (WT) cell lines, each performed in triplicate, normalized to control viability and expressed as percentage specific viability. (B) Data from panel A were used to calculate IC50s. Darker shaded symbols represent cell lines used for subsequent in vitro experimentation. Crossed circles denote the AH29 lymphoma cell line, triangles denote AF47, and squares denote Eµ# 4. (C) Eμ-myc lymphoma cell lines (n = 3), pretreated with 25 μM qVD or a DMSO vehicle control for 1 hour, were exposed to 1000 nM PLX5, 100 nM PLX2, or an additional vehicle control (Con) and viability assessed as in panel A. (D-E) WT, control (pMIH), or BCL-2/BCL-XL transduced Eμ-myc lymphoma cell lines treated with 1000 nM PLX5, 100 nM PLX2, or a DMSO vehicle control (Con) were assessed for viability (D) and changes in mitochondrial membrane potential (Δψm) (E) by annexin V/PI or DiOC6/PI staining, respectively. Points in panels C-E represent the mean of 3 independent experiments for each cell line, performed in triplicate. (F) Matched WT or BCL-2–transduced Eμ-myc lymphoma-bearing C57BL/6 were treated with (n = 7) 10 mg/kg PLX5, (n = 6) 5 mg/kg PLX2, or (n = 11) vehicle control, starting 4 days after engraftment and survival was assessed. Data are collated from 2 independent experiments using 2 different matched pairs of WT and BCL-2–transduced Eμ-myc lymphoma cell lines. Error bars represent standard error of the mean (SEM). Statistical analyses were performed using 1-way (B) or 2-way analysis of variance (ANOVA) (A and C-E) and P values adjusted using Tukey’s or Dunnett’s multiple comparisons tests. In panel A, asterisks represent comparisons of overall curves between different conditions. Survival analysis was performed using a log-rank test adjusted for multiple comparisons using the Holm-Sidak method. ****P < .00005, ***P < .0005, **P < .005, *P < .05. ns, nonsignificant.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/14/10.1182_bloodadvances.2020002231/2/m_advancesadv2020002231f1.png?Expires=1765033668&Signature=P2fIcEAJudRqZ7J5ZKy4dpcQi6~8QTa-RO~b1nN3DSiH-7ksBiLcEThCrxkFalfesR4j7dRLB9xsUWQyTPEbQOu6edBQGSxnq7TIqavddD9FhDTVJI8iVlRJ0QXDMvXRHnL3u78yUtyc3iqwkKqpr8M8zDd1ZF7xJiLstto5OzVndsskOoLf0-tCibDZEYbTnIhKZU9zNNZ87supW~ZQxTsbQ-ZHpOKI-YEbXISTsXAQKG3tUW0wphU6zlhyv6kul6A8rYTmWOUKJHwLzFEGEli4lhsI7D~arMsjeDHPXUByUrRY81y4BX~6TNAvrX9cRU0RYOLLf84PbWKfqOzQqA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
PLX51107 and PLX2853 BETi invoke cell death via intrinsic apoptosis in Eμ-myc lymphomas. (A) PLX51107 (PLX5), PLX2853 (PLX2), JQ1, and OTX015 were administered to Eμ-myc lymphoma cell lines for 48 hours, and viability was assessed by annexin V/PI staining vs vehicle control (dimethyl sulfoxide [DMSO]). Points represent the mean of 12 wild-type (WT) cell lines, each performed in triplicate, normalized to control viability and expressed as percentage specific viability. (B) Data from panel A were used to calculate IC50s. Darker shaded symbols represent cell lines used for subsequent in vitro experimentation. Crossed circles denote the AH29 lymphoma cell line, triangles denote AF47, and squares denote Eµ# 4. (C) Eμ-myc lymphoma cell lines (n = 3), pretreated with 25 μM qVD or a DMSO vehicle control for 1 hour, were exposed to 1000 nM PLX5, 100 nM PLX2, or an additional vehicle control (Con) and viability assessed as in panel A. (D-E) WT, control (pMIH), or BCL-2/BCL-XL transduced Eμ-myc lymphoma cell lines treated with 1000 nM PLX5, 100 nM PLX2, or a DMSO vehicle control (Con) were assessed for viability (D) and changes in mitochondrial membrane potential (Δψm) (E) by annexin V/PI or DiOC6/PI staining, respectively. Points in panels C-E represent the mean of 3 independent experiments for each cell line, performed in triplicate. (F) Matched WT or BCL-2–transduced Eμ-myc lymphoma-bearing C57BL/6 were treated with (n = 7) 10 mg/kg PLX5, (n = 6) 5 mg/kg PLX2, or (n = 11) vehicle control, starting 4 days after engraftment and survival was assessed. Data are collated from 2 independent experiments using 2 different matched pairs of WT and BCL-2–transduced Eμ-myc lymphoma cell lines. Error bars represent standard error of the mean (SEM). Statistical analyses were performed using 1-way (B) or 2-way analysis of variance (ANOVA) (A and C-E) and P values adjusted using Tukey’s or Dunnett’s multiple comparisons tests. In panel A, asterisks represent comparisons of overall curves between different conditions. Survival analysis was performed using a log-rank test adjusted for multiple comparisons using the Holm-Sidak method. ****P < .00005, ***P < .0005, **P < .005, *P < .05. ns, nonsignificant.
PLX51107 and PLX2853 BETi invoke cell death via intrinsic apoptosis in Eμ-myc lymphomas. (A) PLX51107 (PLX5), PLX2853 (PLX2), JQ1, and OTX015 were administered to Eμ-myc lymphoma cell lines for 48 hours, and viability was assessed by annexin V/PI staining vs vehicle control (dimethyl sulfoxide [DMSO]). Points represent the mean of 12 wild-type (WT) cell lines, each performed in triplicate, normalized to control viability and expressed as percentage specific viability. (B) Data from panel A were used to calculate IC50s. Darker shaded symbols represent cell lines used for subsequent in vitro experimentation. Crossed circles denote the AH29 lymphoma cell line, triangles denote AF47, and squares denote Eµ# 4. (C) Eμ-myc lymphoma cell lines (n = 3), pretreated with 25 μM qVD or a DMSO vehicle control for 1 hour, were exposed to 1000 nM PLX5, 100 nM PLX2, or an additional vehicle control (Con) and viability assessed as in panel A. (D-E) WT, control (pMIH), or BCL-2/BCL-XL transduced Eμ-myc lymphoma cell lines treated with 1000 nM PLX5, 100 nM PLX2, or a DMSO vehicle control (Con) were assessed for viability (D) and changes in mitochondrial membrane potential (Δψm) (E) by annexin V/PI or DiOC6/PI staining, respectively. Points in panels C-E represent the mean of 3 independent experiments for each cell line, performed in triplicate. (F) Matched WT or BCL-2–transduced Eμ-myc lymphoma-bearing C57BL/6 were treated with (n = 7) 10 mg/kg PLX5, (n = 6) 5 mg/kg PLX2, or (n = 11) vehicle control, starting 4 days after engraftment and survival was assessed. Data are collated from 2 independent experiments using 2 different matched pairs of WT and BCL-2–transduced Eμ-myc lymphoma cell lines. Error bars represent standard error of the mean (SEM). Statistical analyses were performed using 1-way (B) or 2-way analysis of variance (ANOVA) (A and C-E) and P values adjusted using Tukey’s or Dunnett’s multiple comparisons tests. In panel A, asterisks represent comparisons of overall curves between different conditions. Survival analysis was performed using a log-rank test adjusted for multiple comparisons using the Holm-Sidak method. ****P < .00005, ***P < .0005, **P < .005, *P < .05. ns, nonsignificant.
PLX51107 and PLX2853 upregulate the BH3-only protein BIM in Eμ-myc lymphoma cell lines
To identify key molecular regulators of nonbenzodiazepine BETi-induced apoptosis, BCL-2 family member expression was examined after PLX51107 or PLX2853 application. Increased expression of the proapoptotic BH3-only protein BIM was observed at both mRNA and protein levels in wild-type Eμ-myc LCLs (Figure 2A-C), with PLX2853-induced increases evident at lower concentrations in 3 of 3 cell lines. Increased BIM protein expression was also observed in BCL-2 OE Eμ-myc LCLs (Figure 2D, left; supplemental Figure 2C). Both agents increased association between BIM and BCL-2, representing enhanced mitochondrial priming, in both BCL-2 OE and wild-type Eμ-myc LCLs (Figure 2D-E; supplemental Figure 2D). PUMA and BMF transcripts were also elevated in response to BETi; however, these were not reflected at the protein level (supplemental Figure 4A-B). BCL-2/BCL-XL expression appeared only minimally affected by BETi, although reduced BCL-XL was apparent in 1 cell line. In contrast, both MCL-1 and BCL-2A1 transcripts were significantly upregulated (supplemental Figure 4A). Previously, BCL-2A1/BFL-1 was identified as a critical driver of ABT-199/venetoclax resistance in DLBCL cell lines that was downregulated by benzodiazepine-derivative BETi.31 However, MCL-1 protein remained unaffected by nonbenzodiazepine BETi, and BCL-2A1 was undetectable (supplemental Figure 4B).
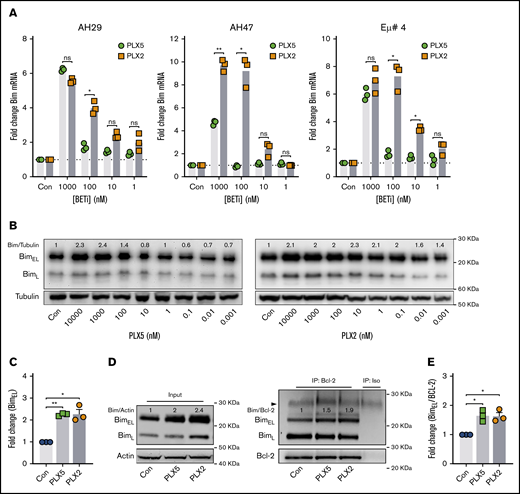
PLX51107 and PLX2853 BETi upregulate the BH3-only protein BIM in Eμ-myc lymphomas. (A) Eμ-myc lymphoma cell lines (AH29, AF47, and Eμ# 4) were treated with qVD (25 μM) for 1 hour to inhibit cell death, followed by PLX5, PLX2, or a DMSO vehicle control (Con) for 24 hours. BIM mRNA expression was then assessed, in triplicate, by quantitative polymerase chain reaction (qPCR) normalized to GAPDH. (B) Eμ-myc lymphoma cell lines (n = 3) were treated as in panel A, and BIM protein expression was assessed after 48 hours. (B) Representative example, with densitometry data normalized to a DMSO control (Con) for 1000 nM PLX5 or PLX2 from each cell line depicted in panel C. (D-E) BCL-2 OE Eμ-myc lymphoma cell lines (n = 3) were treated with 1000 nM PLX5 or PLX2 or a DMSO vehicle control (Con), and BCL-2 protein was immunoprecipitated after 48 hours. Levels of BIM coimmunoprecipitation were then assessed. (D) Representative example of input whole cell lysates (i) and BIM:BCL-2 coimmunoprecipitation (ii) with coimmunoprecipitation densitometry data normalized to baseline demonstrated in panel E. Error bars represent SEM. Statistical analysis was performed using 2-way (A) or 1-way (C-E) ANOVA, and P values were adjusted using the Sidak method or Dunnett’s multiple comparisons tests, respectively. **P < .005, *P < .05.
PLX51107 and PLX2853 BETi upregulate the BH3-only protein BIM in Eμ-myc lymphomas. (A) Eμ-myc lymphoma cell lines (AH29, AF47, and Eμ# 4) were treated with qVD (25 μM) for 1 hour to inhibit cell death, followed by PLX5, PLX2, or a DMSO vehicle control (Con) for 24 hours. BIM mRNA expression was then assessed, in triplicate, by quantitative polymerase chain reaction (qPCR) normalized to GAPDH. (B) Eμ-myc lymphoma cell lines (n = 3) were treated as in panel A, and BIM protein expression was assessed after 48 hours. (B) Representative example, with densitometry data normalized to a DMSO control (Con) for 1000 nM PLX5 or PLX2 from each cell line depicted in panel C. (D-E) BCL-2 OE Eμ-myc lymphoma cell lines (n = 3) were treated with 1000 nM PLX5 or PLX2 or a DMSO vehicle control (Con), and BCL-2 protein was immunoprecipitated after 48 hours. Levels of BIM coimmunoprecipitation were then assessed. (D) Representative example of input whole cell lysates (i) and BIM:BCL-2 coimmunoprecipitation (ii) with coimmunoprecipitation densitometry data normalized to baseline demonstrated in panel E. Error bars represent SEM. Statistical analysis was performed using 2-way (A) or 1-way (C-E) ANOVA, and P values were adjusted using the Sidak method or Dunnett’s multiple comparisons tests, respectively. **P < .005, *P < .05.
PLX51107 and PLX2853 invoke BIM-dependent apoptosis in Eμ-myc lymphoma cell lines
To test the relevance of impaired MYC target transactivation after BETi and BRD4 loading of its promoter,15 the effect of PLX51107 and PLX2853 on the miR-17∼92 miRNA cluster was assessed. miR-17∼92 represents a key target of MYC, required for repression of tumor suppressors (including BIM) to elicit the oncogenic program.32 Both PLX51107 and PLX2853 robustly downregulated the BIM targeting33-36 miRNAs miR-17-5p and -92a (Figure 3A). Overall, a trend toward reduced expression was evident across the miR-17∼92 locus, although because of cell line variation, this was not statistically significant in most cases (supplemental Figure 4C). To determine the contribution of BIM toward PLX51107- and PLX2853-induced apoptosis, BIM-targeted RNAi and BIM−/− (KO) Eμ-myc LCLs were used. BIM shRNA generated ∼75% knockdown (KD) of expression (supplemental Figure 4D). PLX51107- and PLX2853-induced apoptosis appeared unaffected by control transduction (supplemental Figure 4E) but was substantially reduced after BIM KD (Figure 3B). As confirmation, BIM−/− Eμ-myc LCLs, derived from Bim−/− Eμ-myc Tg mice,37 were assessed and an analogous reduction in BETi-induced cell death observed (supplemental Figure 4F). Despite this, BIM KO/KD failed to fully ablate PLX51107- and PLX2853-induced apoptosis, unlike exogenous BCL-2 or BCL-XL expression (Figures 1D and 3B; supplemental Figure 5). Consequently, additional proapoptotic BCL-2 family members are likely to exert an effect. Despite this, Eμ-myc LCLs derived from PUMA−/−, BMF−/−, NOXA−/−, or BIK−/− animals generated in Happo et al38 and Michalak et al,39 exhibited analogous PLX51107- or PLX2853-induced apoptosis to their wild-type counterparts, indicating that a more complex mechanism may generate additional cytotoxicity (supplemental Figure 5). Collectively, these observations identify BIM as a major driver of BETi-induced apoptosis.
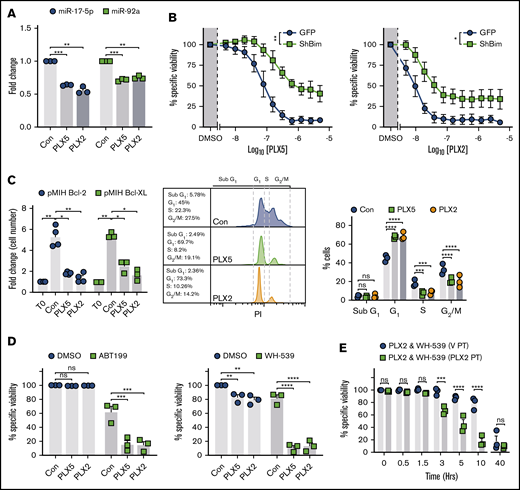
PLX51107 and PLX2853 BETi-induced apoptosis is BIM dependent and effectively combines with BH3-mimetics in Eμ-myc lymphomas. (A) Eμ-myc lymphoma cell lines (n = 3) were treated as in Figure 2A and subjected to 1000 nM PLX5, 100 nM PLX2, or vehicle control (Con) for 24 hours. miR-17-5p and -92a expression was then assessed in triplicate by qPCR normalized to U6 snRNA. (B) Empty vector (GFP) or shBIM-transduced Eμ-myc lymphoma cell lines (n = 3) were treated with PLX5 or PLX2 and assessed for viability as in Figure 1A. Data represent the average of 3 independent experiments for each cell line, performed in triplicate. (Ci) Proliferation of BCL-2– (n = 4) or BCL-XL–transduced (n = 3) Eμ-myc lymphoma cell lines treated with 1000 nM PLX5, 100 nM PLX2, or a DMSO vehicle control (Con) were assessed after 48 hours and depicted as fold change in cell count compared with time 0 (T0). (Cii-iii) BCL-2–transduced (n = 3) Eμ-myc lymphoma cell lines were treated as in panel i and subjected to cell cycle analysis by hypotonic PI staining. (ii) Representative example of hypotonic PI staining. (D) BCL-2– (i) or BCL-XL–OE (ii) Eμ-myc lymphoma cell lines were treated with 1000 nM PLX5, 100 nM PLX2, or an appropriate vehicle control (Con) alongside 300 nM ABT199 (i), 10 μM WEHI-539 (WH-539) (ii), or an additional vehicle control (DMSO) for 48 hours, and viability was assessed by annexin V/PI staining. Points represent the average of 3 independent experiments, performed in triplicate, for each cell line. (E) BCL-XL–transduced Eμ-myc lymphoma cell lines (n = 3) were pretreated with 100 nM PLX2 (PLX2 PT) or a vehicle control (V PT) for 24 hours before administration of 10 μM WEHI-539 (WH-539) and 100 nM PLX2, and viability was assessed after 48 hours. Final PLX2 concentration was 100 nM in all cases. Each point represents the average of triplicate data for each cell line. Statistical analyses were performed via 2-way ANOVA. P values were adjusted for multiple comparisons using either Dunnett’s or Sidak’s analyses, where appropriate, according to experimental design. ****P < .00005, ***P < .0005, **P < .005, *P < .05. Asterisks in panel B represent overall comparisons between curves. Error bars represent SEM.
PLX51107 and PLX2853 BETi-induced apoptosis is BIM dependent and effectively combines with BH3-mimetics in Eμ-myc lymphomas. (A) Eμ-myc lymphoma cell lines (n = 3) were treated as in Figure 2A and subjected to 1000 nM PLX5, 100 nM PLX2, or vehicle control (Con) for 24 hours. miR-17-5p and -92a expression was then assessed in triplicate by qPCR normalized to U6 snRNA. (B) Empty vector (GFP) or shBIM-transduced Eμ-myc lymphoma cell lines (n = 3) were treated with PLX5 or PLX2 and assessed for viability as in Figure 1A. Data represent the average of 3 independent experiments for each cell line, performed in triplicate. (Ci) Proliferation of BCL-2– (n = 4) or BCL-XL–transduced (n = 3) Eμ-myc lymphoma cell lines treated with 1000 nM PLX5, 100 nM PLX2, or a DMSO vehicle control (Con) were assessed after 48 hours and depicted as fold change in cell count compared with time 0 (T0). (Cii-iii) BCL-2–transduced (n = 3) Eμ-myc lymphoma cell lines were treated as in panel i and subjected to cell cycle analysis by hypotonic PI staining. (ii) Representative example of hypotonic PI staining. (D) BCL-2– (i) or BCL-XL–OE (ii) Eμ-myc lymphoma cell lines were treated with 1000 nM PLX5, 100 nM PLX2, or an appropriate vehicle control (Con) alongside 300 nM ABT199 (i), 10 μM WEHI-539 (WH-539) (ii), or an additional vehicle control (DMSO) for 48 hours, and viability was assessed by annexin V/PI staining. Points represent the average of 3 independent experiments, performed in triplicate, for each cell line. (E) BCL-XL–transduced Eμ-myc lymphoma cell lines (n = 3) were pretreated with 100 nM PLX2 (PLX2 PT) or a vehicle control (V PT) for 24 hours before administration of 10 μM WEHI-539 (WH-539) and 100 nM PLX2, and viability was assessed after 48 hours. Final PLX2 concentration was 100 nM in all cases. Each point represents the average of triplicate data for each cell line. Statistical analyses were performed via 2-way ANOVA. P values were adjusted for multiple comparisons using either Dunnett’s or Sidak’s analyses, where appropriate, according to experimental design. ****P < .00005, ***P < .0005, **P < .005, *P < .05. Asterisks in panel B represent overall comparisons between curves. Error bars represent SEM.
BH3 mimetics resensitize BCL-2 or BCL-XL high Eμ-myc lymphoma cell lines to BETi-induced cell death
These data imply that BETi monotherapy is unlikely to be effective in the treatment of B-cell malignancies with high prosurvival BCL-2 family member expression (eg, DEL and most DHL DLBCL) because of their enhanced capacity to neutralize BIM. To identify potential sensitization strategies, BCL-2 or BCL-XL OE Eμ-myc LCLs were used to model these treatment resistant malignancies. Despite lacking apoptotic and therapeutic responses, BCL-2 or BCL-XL OE Eμ-myc LCLs exhibited reduced proliferation, G1 cell cycle arrest, reduced S- and G2/M-phase entry, and elevated BIM expression in response to BETi (Figures 3C and 2D; supplemental Figure 2C). Because both PLX51107 and PLX2853 evoke enhanced BIM-mediated mitochondrial priming (Figure 2D-E; supplemental Figure 2D), we reasoned that coapplication of an appropriate BH3 mimetic (ABT199 for BCL-2 OE, WEHI-53940 for BCL-XL OE) would provoke potent cell death. As expected, exogenous BCL-2 expression sensitized cells to the BCL-2–selective BH3 mimetic ABT199 because of stabilization of BIM expression and increased BIM:BCL-2 association41,42 ; however, this was not reflected by BCL-XL OE after WEHI-539–mediated BCL-XL inhibition (supplemental Figure 3E-F). Accordingly, coadministration of BH3 mimetics alongside a BETi effectively resensitized BCL-2/BCL-XL OE cells to BETi-induced cell death (Figure 3D). Because this combination effect likely requires BETi-mediated BIM upregulation to propagate the effect of BH3 mimetics, prior BETi application may accelerate cell death on BH3 mimetic exposure. An additional 24-hour PLX2853 pretreatment (72 hours total) did not enhance BETi-induced cytotoxicity in comparison with vehicle pretreatment (24 hours of vehicle followed by 48 hours of PLX2853; supplemental Figure 6A-B). However, a more rapid induction of cell death was indeed observed on BH3 mimetic application in comparison with simultaneous compound administration, with responses converging after 48 hours. This was apparent for WEHI-539 (Figure 3E) and, to a lesser extent, ABT199-containing combinations (supplemental Figure 6C). Consequently, appropriate BETi:BH3 mimetic combinations may represent an effective therapeutic approach for MYC-driven BCL-2–high malignancies, such as DEL and most DHL DLBCL.
BETi:BH3 mimetic combinations are synergistic in MYC- and BCL-2–activated DLBCL cell lines
To explore the use of nonbenzodiazepine BETi:BH3 mimetic combinations in human cells, we performed similar experiments in human MYC- and BCL-2–activated GCB DLBCL cell lines (supplemental Table 1). Their relative BIM, BCL-2, and MYC expression profiles are exemplified in supplemental Figure 7A. As expected, MYC- and BCL-2–activated DLBCL cell lines were resistant to BETi-induced cell death (Figure 4A), likely attributable to the increased BIM neutralization capacity afforded by high BCL-2 expression. Consistent with this, PLX51107 and PLX2853 increased BIM protein expression and BCL-2 association (mitochondrial priming) in all cell lines analyzed (Figure 4B-C; supplemental Figure 7B-D). Consequently, resistance to BETi-mediated apoptosis is not attributable to lack of BIM upregulation. Furthermore, human Burkitt lymphoma–derived cell lines, which commonly carry MYC but not BCL-2 translocations, consistently demonstrated elevated BIM protein after PLX51107 or PLX2853 treatment (supplemental Figure 8). BETi-induced BIM upregulation correlated with suppression of miR-17-5p and -92a (Figure 4D) with a variable nonstatistically significant trend toward reduction of other miR-17∼92 constituents (supplemental Figure 7E). Collectively, these data reflect our findings in murine B-lymphoma models. Correspondingly, BETi:ABT199 combinations were synergistic in MYC- and BCL-2–activated DLBCL cell lines, as reported previously for benzodiazepine-derivative BETi31,43,44 (Figure 5A-B; supplemental Figure 9A-B). Consequently, high BCL-2 expression represents a key mediator of BETi-resistance in these cells.
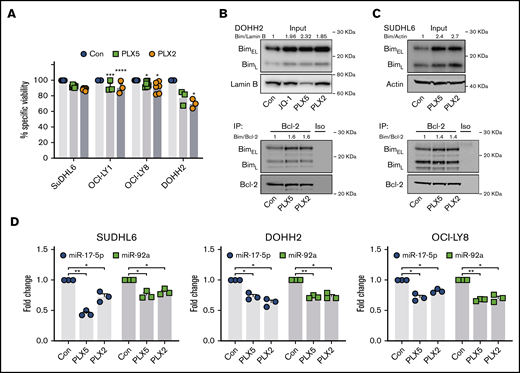
PLX51107 and PLX2853 BETi upregulate BIM in DHL/DEL DLBCL cell lines. (A) SUDHL6, OCI-LY1, OCI-LY8, and DOHH2 were treated with 1000 nM PLX5, 100 nM PLX2, or a DMSO vehicle control (Con) for 48 hours, and viability was assessed by annexin V/PI staining. Points depict the averages of 3 to 6 independent experiments per cell line, each performed in triplicate. (B-C) DOHH2 or SUDHL6 were treated as in panel A, and BIM protein expression was assessed (i). Lysates were subjected to BCL-2 immunoprecipitation and assessed for BIM coimmunoprecipitation (ii). (D) SUDHL6, DOHH2, and OCI-LY8 were treated with 1000 nM PLX5, 100 nM PLX2, or DMSO vehicle control (Con) for 24 hours, and miR-17-5p and -92a expression was assessed by qPCR in triplicate. U6 snRNA was used as an internal control. Error bars represent SEM. Statistical analyses were performed by 2-way ANOVA, and P values were adjusted using Dunnett’s multiple comparisons tests. ****P < .00005, ***P < .0005, **P < .005, *P < .05.
PLX51107 and PLX2853 BETi upregulate BIM in DHL/DEL DLBCL cell lines. (A) SUDHL6, OCI-LY1, OCI-LY8, and DOHH2 were treated with 1000 nM PLX5, 100 nM PLX2, or a DMSO vehicle control (Con) for 48 hours, and viability was assessed by annexin V/PI staining. Points depict the averages of 3 to 6 independent experiments per cell line, each performed in triplicate. (B-C) DOHH2 or SUDHL6 were treated as in panel A, and BIM protein expression was assessed (i). Lysates were subjected to BCL-2 immunoprecipitation and assessed for BIM coimmunoprecipitation (ii). (D) SUDHL6, DOHH2, and OCI-LY8 were treated with 1000 nM PLX5, 100 nM PLX2, or DMSO vehicle control (Con) for 24 hours, and miR-17-5p and -92a expression was assessed by qPCR in triplicate. U6 snRNA was used as an internal control. Error bars represent SEM. Statistical analyses were performed by 2-way ANOVA, and P values were adjusted using Dunnett’s multiple comparisons tests. ****P < .00005, ***P < .0005, **P < .005, *P < .05.
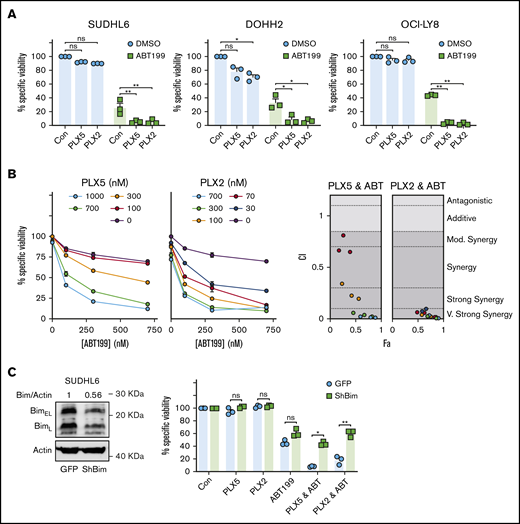
PLX51107 and PLX2853 BETi effectively combine with ABT199 in DHL/DEL cell lines. (A) SUDHL6, DOHH2, and OCI-LY-8 were treated with 1000 nM PLX5, 100 nM PLX2, or a vehicle control (Con) alongside 600 nM ABT199 or an additional vehicle control (DMSO) for 48 hours, and viability was assessed by annexin V/PI staining. (Bi) SUDHL6 was subjected to a dose titration of PLX5 or PLX2 in the presence of increasing ABT199 concentrations and assessed for viability after 48 hours by annexin V/PI staining. Doses (0 nM) represent the addition of an appropriate DMSO vehicle control. (Bii) SUDHL6 data were assessed for synergy using the Chou-Talalay method and CompuSyn software. Points are color coded according to PLX5/PLX2 dose depicted in panel i. Data represent the average of triplicate data. (C) SUDHL6 were transduced with empty vector (GFP) or shBIM constructs, and BIM protein expression was assessed by western blotting (i). The impact of BIM knockdown on the combination of BETi and ABT199 was then assessed by treating SUDHL6 GFP/shBIM as described in panel A. Error bars represent SEM. Statistical analyses were performed using 2-way ANOVA and Dunnett’s multiple comparison tests. **P < .005, *P < .05. Fa, effect; CI, combination index.
PLX51107 and PLX2853 BETi effectively combine with ABT199 in DHL/DEL cell lines. (A) SUDHL6, DOHH2, and OCI-LY-8 were treated with 1000 nM PLX5, 100 nM PLX2, or a vehicle control (Con) alongside 600 nM ABT199 or an additional vehicle control (DMSO) for 48 hours, and viability was assessed by annexin V/PI staining. (Bi) SUDHL6 was subjected to a dose titration of PLX5 or PLX2 in the presence of increasing ABT199 concentrations and assessed for viability after 48 hours by annexin V/PI staining. Doses (0 nM) represent the addition of an appropriate DMSO vehicle control. (Bii) SUDHL6 data were assessed for synergy using the Chou-Talalay method and CompuSyn software. Points are color coded according to PLX5/PLX2 dose depicted in panel i. Data represent the average of triplicate data. (C) SUDHL6 were transduced with empty vector (GFP) or shBIM constructs, and BIM protein expression was assessed by western blotting (i). The impact of BIM knockdown on the combination of BETi and ABT199 was then assessed by treating SUDHL6 GFP/shBIM as described in panel A. Error bars represent SEM. Statistical analyses were performed using 2-way ANOVA and Dunnett’s multiple comparison tests. **P < .005, *P < .05. Fa, effect; CI, combination index.
To determine the role of BIM in this combination and delineate the mechanism of BCL-2–mediated BETi resistance, SUDHL6 cells were subjected to BIM-targeted RNAi with ∼44% BIM KD achieved (Figure 5C, left). Control transduction did not influence BETi-, ABT199-, or combination-induced cytotoxicity. However, BIM KD effectively ablated the combination effect of BETi:ABT199 (Figure 5C, right), indicating a dependence on BIM in these cells. Furthermore, these observations identify that high BCL-2 expression drives resistance to BET-induced apoptosis via neutralization of BETi-upregulated BIM. As a proof of concept, primary human lymphoma cells were subjected to nonbenzodiazepine BETi:BH3 mimetic combination therapy. Primary lymphomas exhibited higher cell death in response to PLX2853:ABT199 combinations compared with either agent alone (supplemental Figure 9C).
BETi:BH3 mimetic combinations demonstrate effective tumor control in MYC- and BCL-2–activated DLBCL xenografts
To determine the in vivo efficacy of BETi:ABT199 combination therapy, the MYC- and BCL-2–translocated DHL DLBCL cell lines SUDHL6 and OCI-LY8 were engrafted into NSG animals. PLX2853, ABT199, and combinations thereof appeared tolerable with no measurable changes in body weight attributable to treatment (supplemental Figure 10). In these models, PLX2853 monotherapy failed to elicit therapeutic responses at doses effective in wild-type Eμ-myc lymphoma-bearing animals, potentially attributable to BCL-2–mediated neutralization of BETi-upregulated BIM, as predicted from in vitro data. Likewise, ABT199 monotherapy was ineffective in these models, presumably because of suboptimal mitochondrial priming in the absence of further apoptotic stimuli. Accordingly, coadministration of ABT199 alongside BETi circumvented the BCL-2–mediated block in intrinsic apoptosis, effectively resensitizing cells to BETi, resulting in a rapid and durable reduction in tumor volume and increased overall survival (Figure 6A-C).
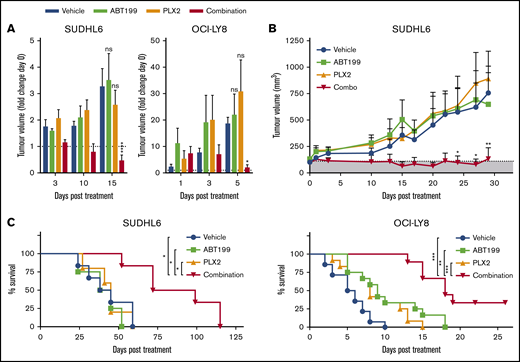
PLX2853 BETi: ABT199 combination therapy enhances survival in DHL xenografts. After detection of tumor growth, SUDHL6- or OCI-LY8–bearing NSG animals were randomized into treatment arms with equivalent mean tumor volume. Subsequently, animals were treated with 5 mg/kg PLX2853 (orally once daily), 50 mg/kg ABT199 (orally once daily), or both in combination, and tumor growth was monitored in comparison with a vehicle control (A-B). Animals were maintained on therapy until the humane end point was reached (OCI-LY8) or for 28 days (SUDHL6), and overall survival was assessed (C). Shaded area in panel B represents tumor size before start of treatment. OCI-LY8 data represent 2 independent experiments combined: vehicle (n = 14), ABT199 (n = 12), PLX2853 (n = 12), combination (n = 9). SUDHL6 data: vehicle (n = 6), ABT199 (n = 4), PLX2853 (n = 5), combination (n = 6). (A-B) Statistical analyses were performed using 2-way ANOVA and Dunnett’s multiple comparisons test. (C) Survival analysis was performed using a log-rank test and Holm-Sidak multiple comparisons correction. Error bars represent SEM. ****P < .00005, ***P < .0005, **P < .005, *P > .05.
PLX2853 BETi: ABT199 combination therapy enhances survival in DHL xenografts. After detection of tumor growth, SUDHL6- or OCI-LY8–bearing NSG animals were randomized into treatment arms with equivalent mean tumor volume. Subsequently, animals were treated with 5 mg/kg PLX2853 (orally once daily), 50 mg/kg ABT199 (orally once daily), or both in combination, and tumor growth was monitored in comparison with a vehicle control (A-B). Animals were maintained on therapy until the humane end point was reached (OCI-LY8) or for 28 days (SUDHL6), and overall survival was assessed (C). Shaded area in panel B represents tumor size before start of treatment. OCI-LY8 data represent 2 independent experiments combined: vehicle (n = 14), ABT199 (n = 12), PLX2853 (n = 12), combination (n = 9). SUDHL6 data: vehicle (n = 6), ABT199 (n = 4), PLX2853 (n = 5), combination (n = 6). (A-B) Statistical analyses were performed using 2-way ANOVA and Dunnett’s multiple comparisons test. (C) Survival analysis was performed using a log-rank test and Holm-Sidak multiple comparisons correction. Error bars represent SEM. ****P < .00005, ***P < .0005, **P < .005, *P > .05.
Collectively, our data demonstrate that nonbenzodiazepine BETi PLX51107 and PLX2853 upregulate the key BH3-only protein BIM, coincident with suppression of BIM-targeting miRNAs, to deliver potent cytotoxic responses and in vivo therapy in BCL-2–low MYC-driven tumors. In contrast, BCL-2–high tumors escape this fate via their increased capacity to bind and neutralize BIM. This mechanistic knowledge can be leveraged to deliver effective in vivo therapy for BCL-2–high MYC-driven tumors via coadministration of an appropriate BH3 mimetic, to deliver durable in vivo tumor control. As such, alteration of the underlying chemical structure of BETi does not alter their therapeutic mechanism.15,29 However, the greater potency of PLX2853 and the potential for reduced toxicity offered by nonbenzodiazepine BETi may allow the clinical potential of BETi to be realized.
Discussion
Because of their ability to modulate the MYC oncogenic network and induce cytotoxicity, BETi represent promising therapeutic agents for B-cell lymphoma.6,11 Despite this, modest responses at the edge of tolerability have limited clinical application of benzodiazepine-derivative BETi in R/R B-lymphoma.18-20
Here we characterized the activities of 2 novel nonbenzodiazepine BETi, PLX5110723 and PLX2853, in murine and human models of MYC-driven lymphoma. PLX51107 and PLX2853 modulated BET biomarker expression (MYC and HEXIM1), consistent with on-target inhibition,6,28 and exhibited potent cytotoxicity in the nanomolar range in Eμ-myc LCLs. Consistent with benzodiazepine-derivative BETi,15,29 PLX51107 and PLX2853 upregulated the key BH3-only protein BIM, coincident with repression of miR-17∼92 cluster constituents miR-17-5p and -92a, leading to BIM-dependent intrinsic apoptosis and in vivo therapeutic responses. In particular, PLX2853 demonstrated elevated potency in comparison with PLX51107 (and benzodiazepine-derivative BETi) in numerous experimental readouts.
BIM represents a critical apoptotic driver during cytotoxic cellular insult, oncogenesis, and development and is central to B-cell homeostasis.37,45,46 This activity is manifest in deletion of mature B cells following disruption of tonic B-cell receptor (BCR) signaling,25 removal of autoreactive B cells,47 and suppression of MYC-driven lymphomagenesis.37 In this latter role, BIM appears to be a direct transcriptional target of MYC.45 However, these activities are repressed by the miR-17∼92 cluster (itself a MYC target).32 In a direct phenocopy of BIM−/− mice,46 forced miR-17∼92 expression drives autoimmunity through BIM suppression, via direct binding of miR-17-5p and -92 to the BIM 3′ untranslated region.33-36 Furthermore, miR-17∼92 KO imparts B-cell deletion associated with elevated BIM expression.35 Consequently, the BIM:miR-17∼92 axis represents a key regulator of B-cell development. However, during MYC-driven oncogenesis, miR-17∼92 facilitates and maintains the MYC-driven neoplastic state through suppression of BIM and epigenetic modifiers.32 These effects appear reversed on experimental MYC inactivation, leading to BIM upregulation- and miR-17∼92 repression-mediated tumor regression.32 This antagonistic relationship appears subverted by the MYC oncogenic program and represents a prime therapeutic target. miR-17∼92 genomic dysregulation is prevalent in GCB DLBCL48 and, where no genomic aberration is apparent, appears driven by upregulation of MYC activity independent of genetics.49 Thus, BIM and miR-17∼92 appear linked to the etiology of MYC-driven tumors.
Benzodiazepine-derivative BETi has been associated with BIM upregulation, ascribed to BETi-mediated removal of BRD4 from the miR-17∼92 promoter.15,29 Here, we expand on these findings and show equivalent effects for the novel nonbenzodiazepine BETi’s PLX51107 and PLX2853. Consequently, our and others’ data demonstrate a direct requirement for BIM induction in BETi-induced apoptosis and in vivo therapy,15 independent of underlying chemical structure. In contrast to experimental MYC inactivation,32 the direct requirement for BETi-mediated MYC downregulation to drive BIM induction has been disputed.15,16 This disparity may arise from enrichment of BRD4 at sites proximal to both MYC and miR-17∼92 promoters.10,50 Consequently, after BETi, both MYC and miR-17∼92 may be independently repressed via displacement of BRD4.
Despite BIM KD or KO affording resistance toward BET-induced apoptosis, protection was not equivalent to that provided by BCL-2 or BCL-XL OE, which fully ablated intrinsic apoptosis (as described previously).15,29 It is likely that additional BH3-only proteins may contribute. BMF and PUMA mRNA were upregulated after BETi. Both BMF and PUMA are implicated in apoptotic tumor suppression after oncogenic MYC dysregulation.39,51 However, neither was robustly upregulated at the protein level, and KO of either gene failed to offer protection to BETi. Potentially, a composite effect resulting from multiple small changes in other BH3-only proteins and/or prosurvival members in different tumors is responsible.
Because exogenous BCL-2 or BCL-XL abrogated cytotoxicity and therapeutic responses in Eμ-myc lymphomas, as described for benzodiazepine-derivative BETi,15,29 PLX51107 or PLX2853 are unlikely to be effective in the treatment of BCL-2–high malignancies,15 such as DEL and most DHL DLBCL.2 Consequently, this may partly explain the disappointing clinical responses seen with benzodiazepine-derivative BETi to date.18 Accordingly, BETi monotherapy was largely ineffective in driving cytotoxic responses and tumor control in MYC- and BCL-2–activated DLBCL cell lines/ primary lymphomas and DLBCL xenografts, respectively. Despite this, BETi-mediated repression of miR-17-5p/-92a and BIM upregulation remained intact. Consequently, neutralization of high BCL-2 expression, via use of the BCL-2–selective BH3 mimetic ABT199/venetoclax, effectively resensitized MYC- and BCL-2–activated DLBCL cell lines and primary lymphomas to BETi-induced cell death and delivered durable antitumor responses in MYC- and BCL-2–translocated DLBCL xenografts. In line with our and others’ data,15,29 this combination effect was BIM dependent. Consequently, enhanced BETi-mediated mitochondrial priming (ie, elevated BIM:BCL-2 association) and subsequent ABT199/venetoclax-enabled displacement likely explains this synergistic effect, as suggested previously for other agents.25,52 BETi:BH3 mimetic combinations may demonstrate particular efficacy in DLBCL, as proposed previously,31 where high BCL-2 expression is common and associated with poor disease-free survival.3
Overall, these findings reflect data obtained using benzodiazepine-derivative BETi both alone and in combination with BH3 mimetics.15,31,53 Consequently, differences in chemical class do not appear to alter the mechanisms of BETi. However, because current benzodiazepine-derivative BETi demonstrate limited clinical responses only at high poorly tolerated doses, the exquisite potency of PLX2853 and the lack of a benzodiazepine backbone (which itself may drive dose-limiting toxicity) may be important for clinical application,18 providing the impetus for further development of PLX2853.
Because our MYC- and BCL-2–activated DLBCL cell lines were exclusively GCB, according to COO classification, the role of BETi-induced BIM upregulation in ABC-DLBCL remains unclear. ABC- and GCB-DLBCL exhibit substantial biological divergence; in particular, reliance on chronic BCR signaling and NF-κB oncogenic addiction in ABC-DLBCL.54 Given the ability of BCR-dependent pathways to regulate BIM expression25 and observations of chronic BCR-signaling in ABC-DLBCL,54 mechanistic differences between COO subtypes may be apparent. Accordingly, NF-κB pathway inhibition has been observed after BETi in ABC DLBCL.55 Presumably, BETi disrupt the BRD4:NF-κB p65 (RELA) interaction, preventing target gene transactivation and formation of new SEs on which ABC-DLBCL rely.54,56
Taken together, our data demonstrate that novel nonbenzodiazepine BETi’s, PLX51107 and PLX2853, potently upregulate the proapoptotic BH3-only protein BIM and repress both MYC and miR-17∼92. These agents drive apoptosis and in vivo therapeutic effects in BCL-2–low MYC-driven lymphomas. In MYC-driven lymphomas where high BCL-2 expression is prevalent (eg, DEL and most DHL DLBCL), coadministration of an appropriate BH3 mimetic resensitizes these cells to therapy and enhances in vivo therapeutic effects via a BIM-dependent mechanism. These combination effects indicate the potential for a viable treatment option in resistant lymphoma.
Authorship
Contribution: T.E.C.C. and M.J.C. performed experiments, analyzed results, and made the figures; M.S.C., P.W.M.J., and M.J.C. designed research; T.E.C.C., M.S.C. and M.J.C. wrote the manuscript with guidance and editing provided by G.P. and P.W.M.J.; Y.M. and B.P. provided biochemical data and identified PLX2853 and PLX51107; K.L.C., T.D.M., A.H.T., L.D., and V.L.E. assisted with in vivo experiments; and R.F. performed STR analysis of cell lines.
Conflict-of-interest disclosure: M.J.C. has, in the past, received funding from Gilead Sciences and Roche. M.S.C. is a retained consultant for Bioinvent and has performed educational and advisory roles for Boehringer Ingelheim, Merck KGaA, Baxalta, and GLG. He has received research funding from Bioinvent, Roche, Gilead, Iteos, UCB, and GSK. Y.M. and B.P. are employees of Plexxikon Inc. The remaining authors declare no competing financial interests.
Correspondence: Matthew J. Carter, Antibody and Vaccine Group, Centre for Cancer Immunology, Cancer Sciences Unit, Faculty of Medicine, University of Southampton, Southampton General Hospital, Tremona Rd, Southampton SO16 6YD, United Kingdom; e-mail: m.j.carter@soton.ac.uk.
Data may be requested from the corresponding author at m.j.carter@soton.ac.uk.
Acknowledgments
The authors acknowledge the patients who contributed samples used in this study. The authors also thank the Experimental Cancer Medicine Centre–funded (C24563/A25171) University of Southampton, Faculty of Medicine Human Tissue Bank (Human Tissue Authority license 12009) for assistance with collecting and characterizing clinical material. Furthermore, the authors acknowledge the contribution of A. Steele for assistance with synergy calculations and Plexxikon Inc. for provision of experimental compounds and guidance in their use. The authors also gratefully acknowledge the contribution of R. Fell and University Hospital Southampton Molecular Pathology department for undertaking cell line STR analysis. In addition, the authors thank A. Egle, L. Happo, E. Michalak, and A. Strasser for provision of BH3-only deficient Eμ-myc LCLs.
This research was supported by a University of Southampton Faculty of Medicine Career Track Fellowship Award to M.J.C. Additional funding was provided by a Bloodwise Specialist Programme (15002) awarded to P.W.M.J., CRUK Centre funding (C328/A25139), and a CRUK Programme grant to G.P. (C2750/A23669).

