

Key Points
Zanubrutinib plus obinutuzumab was safely administered to patients with CLL/SLL and FL, and deep responses were observed.

Abstract
Zanubrutinib (BGB-3111) is a next-generation Bruton tyrosine kinase inhibitor designed to be more selective with fewer off-target effects. We conducted a phase 1 study to assess the safety of its combination with obinutuzumab and evaluate early efficacy in 81 patients with chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL) or relapsed/refractory (R/R) follicular lymphoma (FL). In this phase 1b study, zanubrutinib was tolerable at 160 mg twice daily or 320 mg once daily combined with IV obinutuzumab in patients with CLL/SLL (n = 45) and FL (n = 36). Common adverse events (AEs) included upper respiratory tract infection (51%; n = 23), neutropenia (44%; n = 20), contusion (33%; n = 15), cough, diarrhea, or fatigue (27%; n = 12 each), and pyrexia (22%; n = 10) in CLL/SLL patients and upper respiratory tract infection (39%; n = 14), contusion (28%; n = 10), fatigue (25%; n = 9), and cough (22%; n = 8) in FL patients. Neutropenia was the most common grade 3/4 AE (CLL/SLL, 31% [n = 14]; FL, 14% [n = 5]). Five patients required temporary dose reductions, and 5 discontinued the study drug because of AEs. Overall response rate (ORR) was 100% (n = 20) in treatment-naïve CLL patients and 92% (n = 23) in R/R CLL patients. ORR in 36 R/R FL patients was 72% (n = 26), with 14 complete and 12 partial responses. Median follow-up was 29 months (range, 8-37) for CLL patients and 20 months (range, 2-37) for FL patients. Zanubrutinib and obinutuzumab combination therapy was generally well tolerated. This trial was registered at www.clinicaltrials.gov as #NCT02569476.
Introduction
B-cell receptor signaling is essential for normal B-cell development, but it is also implicated in the survival and proliferation of malignant B cells.1-3 Bruton tyrosine kinase (BTK) is a key component of the B-cell receptor signaling pathway, and the first-generation BTK inhibitor (BTKi) ibrutinib is an established treatment across B-cell malignancies.4
Zanubrutinib (BGB-3111) is a highly specific second-generation BTKi, with favorable oral bioavailability seen in preclinical studies.5,6 Compared with ibrutinib, zanubrutinib has shown greater selectivity for BTK with fewer off-target effects based on multiple in vitro enzymatic and cell-based assays (see supplemental Table 1 in Tam et al7 ). In a recent head-to-head phase 3 comparison in Waldenström macroglobulinemia (WM), zanubrutinib treatment was associated with less toxicity, particularly cardiovascular toxicity, and a trend toward better response quality.8
Zanubrutinib, ibrutinib, and other irreversible BTK inhibitors covalently bind cysteine 481 in the adenosine triphosphate–binding pocket of BTK. They display varying affinities, depending on the specificity of the individual drug, for related and unrelated adenosine triphosphate–binding kinases that contain a sterically available cysteine at this position, including epidermal growth factor receptor (EGFR), human EGFR2 (human epidermal growth factor receptor 2), human EGFR4 (human epidermal growth factor receptor 4), interleukin-2–inducible T-cell kinase (ITK), bone marrow tyrosine kinase gene in chromosome X, Janus-associated kinase 2, Tec, and B-lymphocyte kinase.3,9-11 The relative sparing of ITK by zanubrutinib could result in less interference with the tumor-clearing mechanism of anti-CD20 antibody–induced antibody-dependent cytotoxicity (ADCC), resulting in enhanced efficacy when combined with obinutuzumab.
Toxicities reported in patients treated with ibrutinib include diarrhea and rash specifically associated with EGFR inhibition,12-14 bleeding or bruising,15,16 and atrial fibrillation.17,18 Because these clinical features are not part of the phenotype for males with X-linked agammaglobulinemia (the congenital mutations of the BTK gene preventing production of BTK), it is likely that these adverse effects are due to inhibition of off-target non-BTK kinases rather than inhibition of BTK itself.
Other important features of zanubrutinib include favorable drug-drug interaction characteristics that allow coadministration with azole antifungals or other strong or moderate cytochrome P4503A (CYP3A) inhibitors at a reduced dose,19-21 as well as proton pump inhibitors and antithrombotic agents, including vitamin K antagonists and direct oral anticoagulants, with appropriate monitoring. These characteristics of zanubrutinib were not operative during this phase 1 study but were determined later based on drug-drug interaction studies.
We conducted a phase 1b study of obinutuzumab combined with zanubrutinib to evaluate the safety and tolerability of this chemotherapy-free combination. Here we report the results of cohorts including treatment-naïve (TN) or relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL) and R/R follicular lymphoma (FL).
Patients and methods
Study design
This was a 2-part, open-label, phase 1b clinical trial designed to determine the safety, tolerability, and recommended phase 2 doses (RP2Ds) of zanubrutinib in part 1 and preliminary antitumor activity of zanubrutinib in indication-specific expansion cohorts in part 2. The study was conducted at 15 sites in the United States, Australia, and South Korea and was approved by all institutional review boards and conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. All patients provided written informed consent.
Patients and eligibility criteria
Adult patients with B-cell malignancies CLL/SLL and FL were eligible for enrollment; World Health Organization diagnostic criteria were used.22 Here we report results for patients with CLL/SLL and FL enrolled in part 1 and 2 cohorts. Briefly, patients with TN or R/R CLL/SLL or R/R FL age ≥18 years were eligible. Eastern Cooperative Oncology Group performance status of 0 to 2 and adequate marrow and organ function were required. Prior BTKi exposure was not allowed. Patients were required to have anticipated survival of at least 6 months. Total bilirubin, aspartate aminotransferase, and alanine transaminase were required to be <3× the upper limit of normal. Creatinine clearance ≥30 mL per minute was required. Platelet count of >40 × 109/L and absolute neutrophil count >1.0 × 109/L were required; growth factor use was allowed to bring pretreatment neutrophils to >1.0 × 109/L if marrow infiltration was involved. Patients with R/R FL were not required to have measurable disease, but all 36 enrolled patients had at least 1 nodal lesion >15 mm.
Exclusion criteria included known central nervous system lymphoma or leukemia, prolymphocytic leukemia, history of or currently suspected transformation or Richter syndrome, history of significant heart disease or dysrhythmia, severe pulmonary disease, history of severe allergic/anaphylactic reactions to monoclonal antibody therapy, prior BTKi treatment, ongoing required use of a strong CYP3A inhibitor or inducer, live vaccine treatment within 28 days, allogeneic stem cell transplantation within 6 months or active graft-versus-host disease requiring ongoing immunosuppression, corticosteroids within 7 days of study day 1, chemotherapy or radiotherapy within 3 weeks, monoclonal antibody or any investigational drug within 28 days, other active malignancies within 2 years, compromised gastrointestinal function, major surgery in the past 4 weeks, and active infections (fungal, bacterial, and/or viral infections, including seropositive status for HIV or human T-cell lymphotropic virus).
The study was conducted in 2 parts. Part 1 was a safety evaluation of the combination. Part 2 consisted of indication-specific expansion cohorts and included patients from part 1.
Part 1.
In part 1, patients received zanubrutinib and obinutuzumab until disease progression or intolerable toxicity. The monotherapy RP2Ds had already been established for zanubrutinib in another phase 1 study as either 160 mg twice daily or 320 mg once daily.7 Patients with other B-cell malignancies were eligible for the safety evaluation, and 4 such patients were enrolled (WM, n = 3; marginal zone lymphoma, n = 1); only CLL/SLL and R/R FL patients continued on to part 2. Two dose groups were evaluated for dose-limiting toxicity (DLT): 320 mg once daily and 160 mg twice daily. If ≥2 DLTs were seen in a 6-patient cohort in either regimen, the dose level was considered to have exceeded the maximum tolerated dose, and a reduced dose level of zanubrutinib (160 mg once daily or 80 mg twice daily) would be evaluated in combination with obinutuzumab in another cohort of 6 patients in that regimen. Further reduction of zanubrutinib dose level was allowed until a safe dose combination was identified. The DLT assessment window was 29 days after the first dose. A safety monitoring committee determined dose levels and schedules. Adverse events (AEs) were recorded and graded per National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03).23 A DLT was defined as an AE occurring during the first cycle of treatment, not clearly attributable to a cause other than the study drug and meeting 1 of the following criteria: grade 3/4 nonhematologic toxicity (excluding grade 3 nausea, vomiting, hypertension, and asymptomatic laboratory abnormalities), grade 4 hematologic toxicity persisting for >14 days, and any-grade toxicity requiring removal of the patient from the study.
Part 2.
In part 2, patients from part 1 continued therapy and received zanubrutinib at the doses identified as RP2Ds in part 1: 160 mg twice daily or 320 mg once daily. Additional patients in part 2 were enrolled in indication-specific expansion cohorts for R/R FL, R/R CLL/SLL, and TN CLL/SLL. Cycles lasted 28 days.
In both parts 1 and 2, obinutuzumab was administered IV and concurrently with zanubrutinib in 6 28-day cycles and in accordance with the US label.24 Premedication with glucocorticoid, acetaminophen, and antihistamine was given. On day 1, 100 mg of obinutuzumab was administered over 4 hours, followed on day 2 by 900 mg. On days 8 and 15, 1000 mg was given. On day 1 of cycles 2 to 6, 1000 mg was given. In part 1 only, obinutuzumab infusions were administered 1 day after zanubrutinib.
Treatment modifications
Dosing of zanubrutinib was withheld for patients with select AEs related to the study drug, including grade 4 neutropenia lasting >7 days or grade ≥3 febrile neutropenia, grade 4 thrombocytopenia lasting >7 days or grade ≥3 thrombocytopenia associated with grade ≥2 bleeding, and any-grade ≥3 nonhematologic toxicity except for asymptomatic laboratory abnormalities. Patients could resume treatment if the AE improved to grade ≤1 or baseline within 14 days or within 15 to 28 days; approval from the sponsor was required for the latter.
The restarting dose was kept the same for AEs unrelated to zanubrutinib, but the dose was to be reduced if the AE recurred. For AEs related to zanubrutinib, the restarting dose was reduced. Patients who did not tolerate 80 mg once daily were to be removed from the study. For AEs related to obinutuzumab, this treatment was interrupted or discontinued as medically necessary with no dose modification.
Study end points
Safety and tolerability were the primary end points. Secondary end points included further evaluation of safety, tolerability, and preliminary efficacy. Peripheral blood minimal residual disease (MRD) was also determined for patients with CLL who achieved complete response (CR) where samples could be obtained.
Tumor assessments were performed at the end of weeks 12, 24, 36, and 48, every 24 weeks after week 48, and at disease progression. Physical examination, vital sign, electrocardiogram, and B-symptom assessments were performed at predefined time points. All patients underwent bone marrow examination at screening and at the end of week 12 for those with baseline marrow disease and to confirm any CR. Efficacy was assessed according to International Working Group Criteria for the respective disease25-27 by the investigator at each site; central review was not used.
Statistical analysis
The safety analysis set was also used for efficacy analyses and included all enrolled patients who received ≥1 dose of zanubrutinib or obinutuzumab. This was an early-phase study; therefore, all results are descriptive and provide point estimates and confidence intervals (CIs). Progression-free survival (PFS) and duration of response (DOR) were analyzed by Kaplan-Meier methodology.
Results
Patients
In part 1 of this study, 12 patients were evaluated: 5 with CLL/SLL, 3 with FL, 3 with WM, and 1 with marginal zone lymphoma. In part 2, 40 additional patients with CLL/SLL and 33 additional patients with R/R FL resulted in a total of 45 patients with CLL/SLL (TN, n = 20; R/R, n = 25) and 36 with FL (all R/R). Patient disposition is shown in Figure 1.

Patient disposition. PD, progressive disease; Pt, patient; TN, treatment naïve.
Patient disposition. PD, progressive disease; Pt, patient; TN, treatment naïve.
Median age was 68 years (range, 38-82) for patients with CLL/SLL and 59 years (range, 34-86) for patients with FL. Among patients with CLL/SLL, 39 underwent cytogenetic and molecular analyses that identified 49% (n = 19) with unmutated IGHV, 41% (n = 16) with del(17p)/p53 mutation, 26% (n = 10) with del(11q), and 23% (n = 9) with complex karyotypes. Patients with R/R FL had a median of 2 prior lines of therapy (range, 1-9); at least 39% were rituximab refractory in a prior line, and at least 33% were refractory to their last therapy. Because this was a phase 1 study, specific dates of progression on previous therapies were captured only for patients whose best overall response was PD for prior anticancer regimens. This limited our ability to classify all patients with respect to rituximab-refractory status; 22 patients were either not rituximab refractory or had unknown rituximab-refractory status. Most patients had been previously treated with chemoimmunotherapy in at least 1 prior line of therapy. Other characteristics at study entry are listed in Table 1.
Patient and disease characteristics
| Characteristic | Part 1 (N = 12) | Part 2 | |
|---|---|---|---|
| CLL/SLL (n = 45) | R/R FL (n = 36) | ||
| Age, y | |||
| Median | 68 | 68 | 59 |
| Range | 51-86 | 38-82 | 34-86 |
| ECOG PS | |||
| 0 | 9 | 20 (44) | 28 (78) |
| 1 | 2 | 24 (53) | 6 (17) |
| 2 | 1 | 1 (2) | 2 (6) |
| Prior treatment status | |||
| TN | 0 | 20 (44) | 0 |
| R/R | 12 | 25 (56) | 36 (100) |
| Rituximab-refractory status for R/R FL* | |||
| Refractory | — | — | 14 (39) |
| Not refractory or unknown | — | — | 22 (61) |
| Refractory to most recent line of therapy* | |||
| Refractory | — | — | 12 (33) |
| Not refractory or unknown | — | — | 24 (67) |
| Prior therapies for patients with R/R disease† | n = 25 | n = 36 | |
| Median | 2 | 1 | 2 |
| Range | 1-9 | 1-4 | 1-9 |
| Bulky disease, cm | |||
| Node >5 | — | 15 (33) | 17 (47) |
| Node >10 | — | 0 | 4 (11) |
| Molecular risk factors (n = 39) | |||
| Unmutated IGHV | — | 19 (49) | — |
| Del(17p)/p53 mutation | — | 16 (41) | — |
| Del(11q) | — | 10 (26) | — |
| Complex karyotype | — | 9 (23) | — |
| Characteristic | Part 1 (N = 12) | Part 2 | |
|---|---|---|---|
| CLL/SLL (n = 45) | R/R FL (n = 36) | ||
| Age, y | |||
| Median | 68 | 68 | 59 |
| Range | 51-86 | 38-82 | 34-86 |
| ECOG PS | |||
| 0 | 9 | 20 (44) | 28 (78) |
| 1 | 2 | 24 (53) | 6 (17) |
| 2 | 1 | 1 (2) | 2 (6) |
| Prior treatment status | |||
| TN | 0 | 20 (44) | 0 |
| R/R | 12 | 25 (56) | 36 (100) |
| Rituximab-refractory status for R/R FL* | |||
| Refractory | — | — | 14 (39) |
| Not refractory or unknown | — | — | 22 (61) |
| Refractory to most recent line of therapy* | |||
| Refractory | — | — | 12 (33) |
| Not refractory or unknown | — | — | 24 (67) |
| Prior therapies for patients with R/R disease† | n = 25 | n = 36 | |
| Median | 2 | 1 | 2 |
| Range | 1-9 | 1-4 | 1-9 |
| Bulky disease, cm | |||
| Node >5 | — | 15 (33) | 17 (47) |
| Node >10 | — | 0 | 4 (11) |
| Molecular risk factors (n = 39) | |||
| Unmutated IGHV | — | 19 (49) | — |
| Del(17p)/p53 mutation | — | 16 (41) | — |
| Del(11q) | — | 10 (26) | — |
| Complex karyotype | — | 9 (23) | — |
Values are n (%) except as noted.
ECOG PS, Eastern Cooperative Oncology Group performance status; PI3K, phosphoinositide 3-kinase.
Refractoriness was defined per protocol as less than partial response or PD within 6 mo after completion of prior rituximab-containing therapy (single agent or combination). Missing dates for some patients’ prior lines of therapy limited the number of definitely rituximab-refractory or refractory patients identified; where critical dates were missing, we classified patients conservatively as non–rituximab refractory or unknown.
Prior therapies included nucleoside analogs (fludarabine, gemcitabine, cytarabine, methotrexate), alkylating agents (cyclophosphamide, chlorambucil, bendamustine, ifosfamide, carmustine, melphalan, cisplatin, oxaliplatin, carboplatin), anthracyclines (doxorubicin, epirubicin, pirarubicin), topoisomerase inhibitors (etoposide, mitoxantrone), vinca alkyloids (vinorelbine, vincristine), anti-CD20 agents (rituximab, ofatumumab), BCL2 inhibitor (navitoclax), corticosteroids (dexamethasone, prednisone, prednisolone), histone deacetylase inhibitor (panobinostat), immunomodulators (lenalidomide), PI3K inhibitor (idelalisib), and dual-affinity retargeting antibody.
Treatment exposure
Median duration of treatment with zanubrutinib was 29 months (range, 8-37) for patients with CLL/SLL and 20 months (range, 2-37) for patients with R/R FL. All patients received a median of 6 cycles (range, 1-6) of obinutuzumab. The administered obinutuzumab dose intensity as a percentage of planned dose was 97.6% for patients with CLL/SLL and 99.5% for patients with R/R FL.
Part 1 and safety evaluation
During part 1 of the study, dose levels of 320 mg once daily and 160 mg twice daily were evaluated. No DLT was observed at either dose level, and both schedules were considered acceptable. Overall, 66% of patients were treated at 320 mg once daily and 34% were treated at 160 mg twice daily. Cough, neutropenia, upper respiratory tract infection, contusion, dyspnea, fatigue, peripheral swelling, and petechiae occurred ≥3 times as lower-grade AEs in part 1.
Nine of the 12 patients in part 1 had at least 1 grade ≥3 AE. These higher-grade AEs involved several system organ classes, and neutropenia was the only higher-grade AE to occur more than once (Table 2).
AEs (≥15%) for patients in part 1 (N = 12)
| Preferred term | All grades, n (%) | Grade ≥3, n (%) |
|---|---|---|
| Cough | 5 (41.7) | 0 |
| Neutropenia | 5 (41.7) | 2 (16.7) |
| Upper respiratory tract infection | 4 (33.3) | 0 |
| Contusion | 3 (25) | 0 |
| Dyspnea | 3 (25) | 1 (8.3) |
| Fatigue | 3 (25) | 0 |
| Peripheral swelling | 3 (25) | 1 (8.3) |
| Petechiae | 3 (25) | 0 |
| Abdominal distension | 2 (16.7) | 0 |
| Arthralgia | 2 (16.7) | 0 |
| Basal cell carcinoma | 2 (16.7) | 0 |
| Chronic obstructive pulmonary disease | 2 (16.7) | 1 (8.3) |
| Electrocardiogram QT prolonged | 2 (16.7) | 1 (8.3) |
| Epistaxis | 2 (16.7) | 0 |
| Gastroesophageal reflux disease | 2 (16.7) | 0 |
| Nausea | 2 (16.7) | 0 |
| Nephrolithiasis | 2 (16.7) | 1 (8.3) |
| Edema peripheral | 2 (16.7) | 0 |
| Oral herpes | 2 (16.7) | 0 |
| Pruritus | 2 (16.7) | 0 |
| Rhinitis allergic | 2 (16.7) | 0 |
| Sinusitis | 2 (16.7) | 0 |
| Squamous cell carcinoma of skin | 2 (16.7) | 0 |
| Thrombocytopenia | 2 (16.7) | 1 (8.3) |
| Urinary tract infection | 2 (16.7) | 1 (8.3) |
| Preferred term | All grades, n (%) | Grade ≥3, n (%) |
|---|---|---|
| Cough | 5 (41.7) | 0 |
| Neutropenia | 5 (41.7) | 2 (16.7) |
| Upper respiratory tract infection | 4 (33.3) | 0 |
| Contusion | 3 (25) | 0 |
| Dyspnea | 3 (25) | 1 (8.3) |
| Fatigue | 3 (25) | 0 |
| Peripheral swelling | 3 (25) | 1 (8.3) |
| Petechiae | 3 (25) | 0 |
| Abdominal distension | 2 (16.7) | 0 |
| Arthralgia | 2 (16.7) | 0 |
| Basal cell carcinoma | 2 (16.7) | 0 |
| Chronic obstructive pulmonary disease | 2 (16.7) | 1 (8.3) |
| Electrocardiogram QT prolonged | 2 (16.7) | 1 (8.3) |
| Epistaxis | 2 (16.7) | 0 |
| Gastroesophageal reflux disease | 2 (16.7) | 0 |
| Nausea | 2 (16.7) | 0 |
| Nephrolithiasis | 2 (16.7) | 1 (8.3) |
| Edema peripheral | 2 (16.7) | 0 |
| Oral herpes | 2 (16.7) | 0 |
| Pruritus | 2 (16.7) | 0 |
| Rhinitis allergic | 2 (16.7) | 0 |
| Sinusitis | 2 (16.7) | 0 |
| Squamous cell carcinoma of skin | 2 (16.7) | 0 |
| Thrombocytopenia | 2 (16.7) | 1 (8.3) |
| Urinary tract infection | 2 (16.7) | 1 (8.3) |
Safety
The safety analysis set included all CLL/SLL patients from part 1 and all patients from part 2. All patients with CLL/SLL and 97% of patients with FL reported at least 1 AE; 96% (n = 43) and 83% (n = 30) had treatment-related AEs, respectively. The most common AEs in >20% of patients with CLL/SLL included upper respiratory tract infection (51%), neutropenia (44%), contusion (33%), cough, diarrhea, or fatigue (27% each), and pyrexia (22%); patients with FL experienced upper respiratory tract infection (39%), contusion (28%), fatigue (25%), and cough (22%; Table 3). Neutropenia was the most common grade ≥3 AE in both patients with CLL/SLL (31%) and those with R/R FL (14%). A single event of major hemorrhage occurred in this study when a patient with epistaxis had to be hospitalized overnight for observation because of the patient’s remote home address. No atrial fibrillation or flutter was reported in this study. No high-grade diarrhea was reported, and grade ≥3 hypertension was limited to 3 patients with CLL/SLL and 3 with R/R FL (Table 4). Serious AEs were reported in 49% (n = 22) of patients with CLL/SLL and 33% (n = 12) of patients with FL, with pneumonia being the most common. Four (9%) of 45 patients with CLL/SLL had an AE that led to treatment discontinuation of zanubrutinib (grade 2 erythema nodosum, grade 3 disseminated Cryptococcus, grade 5 metastatic squamous cell carcinoma, and grade 3 pneumonia). One (3%) of 36 patients with R/R FL had an AE of grade 2 lethargy also associated with grade 2 neutropenia and grade 3 thrombocytopenia, which led to treatment discontinuation. Five patients had temporary dose reductions during the study, 4 with CLL/SLL and 1 with R/R FL. AEs associated with these dose reductions were maculopapular rash (grade 1), elevated alkaline phosphatase (grade 2 for 3 days), neutropenia (grade 4 for 7 days and grade 2 for 3 days), loss of balance (grade 2 and chronic), and constipation (grade 2 and chronic).
AEs (≥15%) for patients in part 2
| Preferred term | All grades, n (%) | Grade ≥3, n (%) |
|---|---|---|
| CLL/SLL (n = 45) | ||
| Upper respiratory tract infection | 23 (51.1) | 0 |
| Neutropenia | 20 (44.4) | 14 (31.1) |
| Contusion | 15 (33.3) | 0 |
| Cough | 12 (26.7) | 0 |
| Diarrhea | 12 (26.7) | 0 |
| Fatigue | 12 (26.7) | 1 (2.2) |
| Pyrexia | 10 (22.2) | 0 |
| Petechiae | 8 (17.8) | 0 |
| Rash maculopapular | 8 (17.8) | 0 |
| Sinusitis | 8 (17.8) | 0 |
| Thrombocytopenia | 8 (17.8) | 3 (6.7) |
| Back pain | 7 (15.6) | 2 (4.4) |
| Dyspepsia | 7 (15.6) | 0 |
| Nausea | 7 (15.6) | 0 |
| Pneumonia | 7 (15.6) | 4 (8.9) |
| Rash | 7 (15.6) | 0 |
| R/R FL (n = 36) | ||
| Upper respiratory tract infection | 14 (38.9) | 0 |
| Contusion | 10 (27.8) | 0 |
| Fatigue | 9 (25.0) | 0 |
| Cough | 8 (22.2) | 0 |
| Nausea | 7 (19.4) | 0 |
| Rash | 7 (19.4) | 0 |
| Thrombocytopenia | 7 (19.4) | 2 (5.6) |
| Diarrhea | 6 (16.7) | 0 |
| Neutropenia | 6 (16.7) | 5 (13.9) |
| Petechiae | 6 (16.7) | 0 |
| Productive cough | 6 (16.7) | 0 |
| Preferred term | All grades, n (%) | Grade ≥3, n (%) |
|---|---|---|
| CLL/SLL (n = 45) | ||
| Upper respiratory tract infection | 23 (51.1) | 0 |
| Neutropenia | 20 (44.4) | 14 (31.1) |
| Contusion | 15 (33.3) | 0 |
| Cough | 12 (26.7) | 0 |
| Diarrhea | 12 (26.7) | 0 |
| Fatigue | 12 (26.7) | 1 (2.2) |
| Pyrexia | 10 (22.2) | 0 |
| Petechiae | 8 (17.8) | 0 |
| Rash maculopapular | 8 (17.8) | 0 |
| Sinusitis | 8 (17.8) | 0 |
| Thrombocytopenia | 8 (17.8) | 3 (6.7) |
| Back pain | 7 (15.6) | 2 (4.4) |
| Dyspepsia | 7 (15.6) | 0 |
| Nausea | 7 (15.6) | 0 |
| Pneumonia | 7 (15.6) | 4 (8.9) |
| Rash | 7 (15.6) | 0 |
| R/R FL (n = 36) | ||
| Upper respiratory tract infection | 14 (38.9) | 0 |
| Contusion | 10 (27.8) | 0 |
| Fatigue | 9 (25.0) | 0 |
| Cough | 8 (22.2) | 0 |
| Nausea | 7 (19.4) | 0 |
| Rash | 7 (19.4) | 0 |
| Thrombocytopenia | 7 (19.4) | 2 (5.6) |
| Diarrhea | 6 (16.7) | 0 |
| Neutropenia | 6 (16.7) | 5 (13.9) |
| Petechiae | 6 (16.7) | 0 |
| Productive cough | 6 (16.7) | 0 |
AEs of interest
| AE | CLL/SLL, n (%) (n = 45) | FL, n (%) (n = 36) | ||
|---|---|---|---|---|
| All grades | Grade ≥3 | All grades | Grade ≥3 | |
| Infections | 39 (87) | 17 (38) | 24 (67) | 7 (19) |
| Diarrhea | 12 (27) | 0 | 6 (17) | 0 |
| Infusion-related reaction | 11 (24) | 1 (2) | 5 (14) | 0 |
| Hypertension | 4 (9) | 3 (7) | 3 (8) | 3 (8) |
| Major hemorrhage | 0 | 0 | 1 (3) | 0 |
| Atrial fibrillation | 0 | 0 | 0 | 0 |
| AE | CLL/SLL, n (%) (n = 45) | FL, n (%) (n = 36) | ||
|---|---|---|---|---|
| All grades | Grade ≥3 | All grades | Grade ≥3 | |
| Infections | 39 (87) | 17 (38) | 24 (67) | 7 (19) |
| Diarrhea | 12 (27) | 0 | 6 (17) | 0 |
| Infusion-related reaction | 11 (24) | 1 (2) | 5 (14) | 0 |
| Hypertension | 4 (9) | 3 (7) | 3 (8) | 3 (8) |
| Major hemorrhage | 0 | 0 | 1 (3) | 0 |
| Atrial fibrillation | 0 | 0 | 0 | 0 |
Infection was a common overall AE, but most were grade 1/2 upper respiratory infections. Four grade ≥3 pneumonia events occurred in the CLL/SLL population. There were 3 infections occurring in patients with CLL/SLL classified as opportunistic infections: Pneumocystis jirovecii pneumonia, disseminated cryptococcal infection, and listeriosis.
Sixteen of 81 patients (20%) had an infusion reaction with obinutuzumab; of these, 1 was grade ≥3.
Efficacy
At a median follow-up of 29 months (range, 8-37) for patients with TN CLL/SLL, overall response rate (ORR) was 100% (n = 20), including 6 CRs (30%); for patients with R/R CLL/SLL, ORR was 92% (n = 23), including 7 CRs (28%). Of 16 patients with del(17p), 6 (100%) of 6 TN patients and 8 (80%) of 10 R/R patients had an objective response (Table 5), with 50% and 20% achieving CR, respectively. Median DOR by the Kaplan-Meier method was not reached for CLL/SLL.
Disease response
| Response | TN CLL/SLL (n = 20) | R/R CLL/SLL (n = 25) | R/R FL (n = 36) |
|---|---|---|---|
| ORR | 20 (100) | 23 (92) | 26 (72) |
| Best response | |||
| CR | 6 (30) | 7 (28) | 14 (39) |
| PR | 14 (70) | 16 (64) | 12 (33) |
| Stable disease | 0 | 2 (8) | 6 (17) |
| PD | 0 | 0 | 4 (11) |
| ORR for del(17p)/p53 mutation (n = 16) | 6 of 6 (100) | 8 of 10 (80) | — |
| Follow-up period, mo | |||
| Median | 29 | 29 | 20 |
| Range | 14-35 | 8-37 | 2-37 |
| Response | TN CLL/SLL (n = 20) | R/R CLL/SLL (n = 25) | R/R FL (n = 36) |
|---|---|---|---|
| ORR | 20 (100) | 23 (92) | 26 (72) |
| Best response | |||
| CR | 6 (30) | 7 (28) | 14 (39) |
| PR | 14 (70) | 16 (64) | 12 (33) |
| Stable disease | 0 | 2 (8) | 6 (17) |
| PD | 0 | 0 | 4 (11) |
| ORR for del(17p)/p53 mutation (n = 16) | 6 of 6 (100) | 8 of 10 (80) | — |
| Follow-up period, mo | |||
| Median | 29 | 29 | 20 |
| Range | 14-35 | 8-37 | 2-37 |
Values are n (%) except as noted.
PR, partial response.
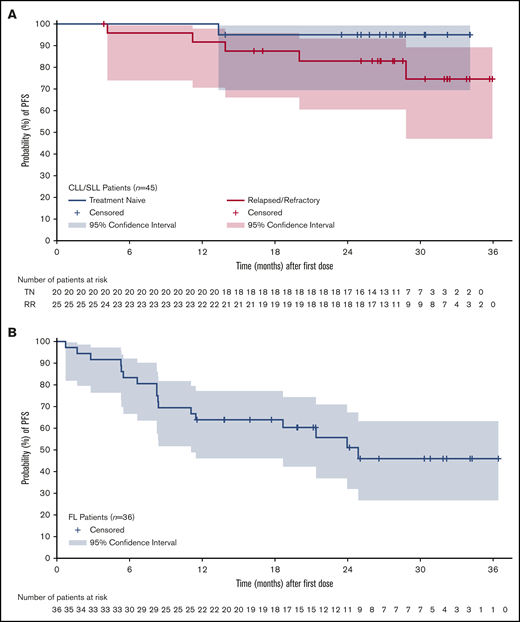
The estimated event-free rate for DOR at 24 months was 90.4% (95% CI, 76.5% to 96.3%). Median PFS was not reached for patients with CLL/SLL (Figure 2A); 73% (n = 33) remained on treatment at the data cutoff date, and 24% (n = 12) had discontinued treatment because of PD (n = 5), AE (n = 4), patient decision (n = 1), investigator decision (n = 1), or other reason (n = 1). Five patients with CLL/SLL died, 4 as a result of PD and 1 as a result of metastatic squamous cell carcinoma, which had been treated with curative intent in the distant past.
Peripheral blood samples from TN CLL/SLL responders with CR (n = 6) were tested by flow cytometry for MRD28 at a median of 23.5 cycles (range, 22-29). Among these, 3 were MRD− at the 10−4 detection limit.
Among patients with R/R FL, at a median follow-up of 20 months (range, 2-37), ORR was 72% (n = 26), with a CR rate of 39% (Table 5). Median DOR by the Kaplan-Meier method was not reached (95% CI, 18.4 months to not estimable). The estimated event-free rates for DOR at 18 and 24 months were 75.5% (95% CI, 53.1% to 88.3%) and 62.3% (95% CI, 37.0% to 79.9%), respectively. Median PFS for the FL patient population was 25 months (range, 0.7-36; Figure 2B). Half of the patients with FL (n = 18) remained on study treatment at the data cutoff date, and the other 50% had discontinued because of PD (n = 14), patient decision (n = 2), AE (n = 1), and investigator decision (n = 1). Four patients with R/R FL died as a result of PD.
Discussion
Anti-CD20 antibodies play a fundamental role in the treatment of patients with B-cell malignancies, with obinutuzumab having proven benefit.29-31 The second-generation BTKi zanubrutinib shows improved selectivity compared with ibrutinib; it is hypothesized to result in lower incidence of toxicities thought to be mediated through inhibition of off-target kinases and also may allow more effective combination with anti-CD20 agents. The relative sparing of ITK by zanubrutinib could result in less interference with the tumor-clearing mechanism of anti-CD20 antibody–induced ADCC, resulting in enhanced efficacy.
In this phase 1b study, the combination of obinutuzumab with zanubrutinib was tolerable at the standard monotherapy doses of 320 mg once daily and 160 mg twice daily. The toxicity profile was broadly consistent with the safety profile observed with zanubrutinib monotherapy,32 with the exception of neutropenia in CLL/SLL patients, which was clearly more common with the combination (44%) reported here than with zanubrutinib monotherapy (7.4%) in the phase 1 study.7 As such, much of the neutropenia observed in this study occurred in patients with CLL/SLL (31% grade ≥3). In the iLLUMINATE study, all-grade neutropenia occurred in 43% of 113 CLL/SLL patients receiving ibrutinib plus obinutuzumab, with grade ≥3 events in 36%.33 Neutropenia was less common among 36 R/R FL patients; 17% had neutropenia of any grade, and 14% had grade ≥3 events. In the GAUSS study, single-agent obinutuzumab in patients with indolent B-cell lymphoma (mostly FL) had a neutropenia rate of 3% (all grades),31 whereas in the GAUGUIN study, the single-agent rate was 13.6%.34
The lower rate of diarrhea observed with zanubrutinib (∼22%), with no grade ≥3 event, is consistent with the relative sparing of EGFR. No atrial fibrillation was reported in the current study, and the incidence of all-grade hypertension was <10%. Low-grade skin bleeding, thought to be an on-target effect of BTK inhibition on platelet function, occurs with zanubrutinib.15,35 Although comparisons across studies are limited, the relative sparing by zanubrutinib of other kinases that are involved in hemostasis, such as Tec, may favorably influence the incidence and severity of bleeding compared with ibrutinib, which has off-target effects that exacerbate the platelet defect.7,36 Regarding long-term tolerability, only 5 (6.2%) of 81 patients discontinued because of an AE. Recently, the first head-to-head comparison of 2 BTK inhibitors was published, where zanubrutinib was directly compared with ibrutinib in 201 patients with WM. Incidence of atrial fibrillation, contusion, diarrhea, peripheral edema, hemorrhage, muscle spasms, and pneumonia, as well as AEs leading to treatment discontinuation, was lower with zanubrutinib.8 As in the current study, neutropenia was higher with zanubrutinib, although grade ≥3 infection rates were similar.
BTK inhibition can mitigate obinutuzumab-induced infusion-related reactions. Recent evidence suggests that this may involve 3 cytokines: CCL3, IFN-γ, and TNF-α, all of which showed a significant increase along with clinical symptoms in patients who developed an obinutuzumab-related infusion reaction.37 In this study, infusion-related reactions were observed in <20% of patients (<3% grade ≥3), compared with a reported incidence of 65% with obinutuzumab monotherapy and consistent with observations for the combination of ibrutinib and obinutuzumab.37
In addition to a tolerable safety profile, the combination of zanubrutinib and obinutuzumab in patients with CLL/SLL showed preliminary efficacy. Anti-CD20 antibodies such as rituximab and obinutuzumab have demonstrated improvements in PFS and OS when combined with standard therapies,38,39 as well as novel agents such as venetoclax.29,40 In the iLLUMINATE study, ibrutinib plus obinutuzumab showed a significant improvement in PFS vs chlorambucil plus obinutuzumab standard chemoimmunotherapy.33 In the 3-arm ELEVATE TN study, at a median follow-up of 28.3 months, acalabrutinib plus obinutuzumab produced a 90% reduction in the risk of PD or death compared with chlorambucil plus obinutuzumab.41
The addition of rituximab to ibrutinib does not seem to improve PFS in patients with CLL, possibly because of interference with ADCC through off-target inhibition of ITK.42 This is supported by ALLIANCE trial data showing that ibrutinib monotherapy and ibrutinib plus rituximab (IR) were associated with superior PFS vs rituximab plus bendamustine in patients age ≥65 years with TN CLL. The 2-year PFS estimates were 74% for rituximab plus bendamustine, 87% for ibrutinib, and 88% for IR.43 These results persisted with longer follow-up in the ALLIANCE trial.44 Another phase 2 trial compared patients with CLL/SLL (N = 208) treated with ibrutinib or IR without prolongation of PFS.42 Although longer follow-up is needed, the lack of separation between the PFS curves supports a preliminary conclusion that the addition of rituximab to ibrutinib does not prolong PFS in patients with CLL/SLL.
Follow-up is still early for PFS in patients with CLL in this study; however, response rates were high (100% for TN patients and 92% for R/R patients). The CR rates (30% in TN patients and 28% in R/R patients) observed also support zanubrutinib as a potentially effective partner with anti-CD20 and may lead to improvement over the robust results with zanubrutinib monotherapy.7
Efficacy of this combination in heavily pretreated R/R FL patients was demonstrated (ORR, 72%), with more than half of responding patients achieving CR. This contrasts with the response rate of 41% for zanubrutinib monotherapy in R/R FL, which was not considered adequate for further clinical development. Responses to the combination were durable (median PFS, 25 months).45 The best reported response rate for obinutuzumab monotherapy was obtained in the GAUSS study (best ORR, 66%, with a 42% CR/unconfirmed CR rate as determined by investigators).27 However, patients in GAUSS were required to have had a documented response lasting ≥6 months after completion of their last rituximab dose in a prior therapy and were less often rituximab refractory (9.4%), compared with at least 39% who were rituximab refractory in this study. Ibrutinib monotherapy in the R/R FL setting has limited efficacy, with the DAWN study showing an ORR of 20.9% and median PFS of just 4.6 months,46 although patients in that study had higher-risk disease (requiring ≥2 prior lines of therapy and progression within 12 months after their most recent line of therapy). Taken together, these results suggest that BTK inhibition alone in R/R FL may not be optimal, and combination therapies should be considered. The role of obinutuzumab in FL has been established in several studies, including both frontline and R/R FL.30,34,47 The preliminary activity in the heavily pretreated patients described here supports further study of this combination.
Recent studies have examined other nonchemotherapy approaches in R/R FL, including lenalidomide and PI3K inhibitors (PI3Kis). In the AUGMENT study of lenalidomide plus rituximab in R/R FL, ORR was 78%, with a CR rate of 34%. However, the AUGMENT study used a favorable-risk population, where 57% had only 1 prior line of therapy, and rituximab-refractory patients were excluded.48 This is in contrast to our study, where at least 14 (39%) of 36 patients had rituximab-refractory disease, and 12 (33%) were refractory to their most recent therapy. Three PI3Kis, idelalisib, duvelisib, and copanlisib, have received accelerated approval in the United States for R/R FL, showing ORRs ranging from 42% to 59% and CRs in ∼15% of patients with R/R FL, with median PFS of <1 year. The safety profile of zanubrutinib plus obinutuzumab is favorable compared with that of PI3Kis, which carry risks of serious infection and immune-related events such as colitis.
In conclusion, our results demonstrate that the combination of zanubrutinib and obinutuzumab seems tolerable in CLL and FL and clinical responses were observed. The study is limited by its preliminary nature, small number of patients, and lack of a control group. The safety results of this trial, particularly the relatively low rate of AEs requiring treatment discontinuation, support continued evaluation of this combination. A global randomized phase 2 study of the combination is currently enrolling patients with R/R FL. For CLL/SLL, alternative novel combinations are being evaluated.
All authors vouch for the accuracy of the study data. The data were analyzed by the study statistician and coauthor at BeiGene (J.S.) and interpreted by the authors. Individual participant data will not be shared before regulatory approval of zanubrutinib for the treatment of CLL/SLL and FL. Requests for copies of the protocol and statistical analysis plan will be considered. Please contact constantine.tam@petermac.org.
Acknowledgments
The authors thank the patients and their families and gratefully acknowledge expert technical and organizational assistance from Katerina Ajami at BeiGene.
This work, including medical writing and editorial assistance, was supported by BeiGene USA, Inc. Writing and editorial support were provided by A. Daisy Goodrich and Bio Connections (Chicago, IL), funded by BeiGene USA, Inc. (San Mateo, CA).
Authorship
Contribution: J.H., R.P., A.C., R.E., W.R., J.S., and C.S.T. were responsible for study design; C.S.T. contributed to data interpretation and analysis; C.S.T., H.Q., A.N., X.B., H.R., H.M.P., M.F.L., R.E., N.W., S.S.P., and I.W.F. and their respective research teams reviewed patient records and contributed to data collection; J.H., R.P., A.C., R.E., W.R., and J.S. confirmed data accuracy; C.S.T. contributed to the first draft of the manuscript, contributed to final manuscript writing, and had final responsibility to submit for publication; and all authors had full access to all of the data, carefully reviewed the manuscript, and approved the final version.
Conflict-of-interest disclosure: C.S.T. received research funding from Janssen, AbbVie, BeiGene, Pharmacyclics, and TG Therapeutics and served as a consultant for BeiGene, Janssen, Roche, AbbVie, and LOXO. H.Q. served as a consultant for Celgene, Janssen Cilag, Takeda, Karyopharm, and Amgen and received research funding from Celgene and Amgen. A.N. received research funding from Parexel and travel funding from Amgen, Janssen, and Novartis. X.B. received honoraria from Roche and served as a consultant for AbbVie. H.M.P. received honoraria from Takeda, Janssen, Amgen, Celgene, and Allergan; served as a consultant for Takeda, Janssen, Amgen, and Allergan; and received research funding from Allergan. M.F.L. received honoraria from Vifor Pharma and travel funding from Amgen. N.W. has equity ownership in Icon Consolidated Holdings and received travel funding from Celgene. J.H. is an employee of, has a leadership role in, and has equity ownership in BeiGene USA. A.C. is an employee of, has equity ownership in, and received travel funding from BeiGene USA. R.E. is an employee of BeiGene and has equity ownership in BeiGene USA and Roche. R.P. is an employee of BeiGene USA and has equity ownership in BeiGene USA and Amgen. W.R. is an employee of, has equity ownership in, and received travel funding from BeiGene USA. J.S. is an employee of and has equity ownership in BeiGene USA. I.W.F. served as a consultant for AbbVie, Seattle Genetics, TG Therapeutics, and Verastem and received research funding from Acerta Pharma, Agios, Calithera Biosciences, Celgene, Constellation Pharmaceuticals, Genentech, Gilead Sciences, Incyte, Infinity Pharmaceuticals, Janssen, Karyopharm Therapeutics, Kite Pharma, Novartis, Pharmacyclics, Portola Pharmaceuticals, Roche, TG Therapeutics, Trilliam Therapeutics, AbbVie, ArQule, BeiGene, Curis, FORMA Therapeutics, Forty Seven, Merck, Pfizer, Takeda, Teva, Verastem, Gilead Sciences, AstraZeneca, Juno Therapeutics, Unum Therapeutics, and MorphoSys. The remaining authors declare no competing financial interests.
Correspondence: Constantine S. Tam, Peter MacCallum Cancer Centre, 305 Grattan St, Melbourne, VIC 3050, Australia; e-mail: constantine.tam@petermac.org.

