

Key Points
Administration of CpG-MSCs to neutropenic mice protects against sepsis by augmenting emergency granulopoiesis through paracrine factors.
Neutrophils from irradiated mice cocultured with CpG-MSCs produce less neutrophil extracellular traps and have less organ damage in sepsis.

Abstract
Patients with immune deficiencies from cancers and associated treatments represent a growing population within the intensive care unit with increased risk of morbidity and mortality from sepsis. Mesenchymal stromal cells (MSCs) are an integral part of the hematopoietic niche and express toll-like receptors, making them candidate cells to sense and translate pathogenic signals into an innate immune response. In this study, we demonstrate that MSCs administered therapeutically in a murine model of radiation-associated neutropenia have dual actions to confer a survival benefit in Pseudomonas aeruginosa pneumo-sepsis that is not from improved bacterial clearance. First, MSCs augment the neutrophil response to infection, an effect that is enhanced when MSCs are preconditioned with CpG oligodeoxynucleotide, a toll-like receptor 9 agonist. Using cytometry by time of flight, we identified proliferating neutrophils (Ly6GlowKi-67+) as the main expanded cell population within the bone marrow. Further analysis revealed that CpG-MSCs expand a lineage restricted progenitor population (Lin−Sca1+C-kit+CD150−CD48+) in the bone marrow, which corresponded to a doubling in the myeloid proliferation and differentiation potential in response to infection compared with control. Despite increased neutrophils, no reduction in organ bacterial count was observed between experimental groups. However, the second effect exerted by CpG-MSCs is to attenuate organ damage, particularly in the lungs. Neutrophils obtained from irradiated mice and cocultured with CpG-MSCs had decreased neutrophil extracellular trap formation, which was associated with decreased citrullinated H3 staining in the lungs of mice given CpG-MSCs in vivo. Thus, this preclinical study provides evidence for the therapeutic potential of MSCs in neutropenic sepsis.
Introduction
Sepsis is a devastating disease caused by a dysregulated immune response to infection associated with multisystem organ failure and high mortality.1 Despite improved clinical care, sepsis contributes to more than 5 million global deaths per year,2 and accounts for $20 billion (5.2%) of total US hospital costs.3 Patients with immune deficiencies, for example, from cancers and their associated treatments, comprise 30.6% of patients admitted to the intensive care unit with severe sepsis and septic shock, and have an increased risk of death.4,5 Patients who have undergone hematopoietic stem cell transplantation (HSCT) represent a particularly vulnerable population, as they have a fivefold higher risk of sepsis compared with those who are immunocompetent.6 In the peri-transplant period, 10% to 20% of HSCT patients will require an admission to the intensive care unit, which is associated with high (44%-70%) mortality.7,8 This is likely due to complicating neutropenia and the adverse effects of myeloablative conditioning regimens.6-9
Total body irradiation, an integral part of many conditioning regimens for hematopoietic stem cell transplantation,10 is associated with ablation and dysfunction of both immune11 and stromal cells.12 This can have deleterious effects on the host’s ability to initiate emergency granulopoiesis,13 the de novo generation of neutrophils in response to a systemic bacterial infection through increased proliferation of myeloid progenitor cells.14-16 Pathogen sensing initiates this process, and nonhematopoietic cells have been shown to be critical.17 Boettcher et al demonstrated that endothelial cells are indispensable drivers of lipopolysaccharide (LPS; toll-like receptor 4-MyD88 [TLR4-MyD88])-induced emergency granulopoiesis, but that alternative redundant pathways exist when mice were challenged with live bacteria.16
Mesenchymal stromal cells (MSCs) are fibroblast-like cells that express TLRs18,19 and contribute to the hematopoietic niche.20 They are defined by in vitro plastic adherence, trilineage (osteoblast, adipocyte, and chondroblast) differentiation potential, negative immune lineage markers, and positive mesenchymal markers.21 Importantly, MSCs have pathogen-sensing ability through TLR signaling and can modulate the bone marrow22 and neutrophil response to infection23 ; thus, they may play an additive role in emergency granulopoiesis. Furthermore, MSCs have been administered to HSCT patients in phase 1/2 clinical trials to promote stem cell engraftment and for the treatment of graft-versus-host disease, and represents a feasible and safe therapeutic option in this patient population.24 The many therapeutic properties of MSCs include the ability to home to inflamed tissues,25 and the facilitation of cellular crosstalk via their rich secretome, which comprise bioactive molecules including lipids, proteins, and nucleic acids that have been able to recapitulate many of the therapeutic effects exerted by whole cells.26 Although MSCs are defined by their in vitro characteristics, their functions can vary significantly depending on their inflammatory microenvironment.27,28 Preconditioning MSCs with cytokines and growth factors can additionally modulate their function and secretory repertoire.29
DNA sequences that contain abundant hypomethylated GC dinucleotides (CpG) are highly enriched in bacterial genomes and are 100-fold less bountiful in eukaryotic DNA, except in the mitochondria.30 These oligonucleotide sequences primarily signal through the endosomal TLR9 pathway,31 although TLR9-independent pathways have also been described.32,33 Stimulation of TLR9 by circulating mitochondrial DNA can act as damage associated molecular patterns and activate innate immune responses independent of the presence of pathogens.34 The effect of CpG sequences, however, can vary depending on the cell type. Notably, treatment of MSCs with oxidized cell-free DNA has been shown to induce adaptive responses to further oxidative stress.35 Given this, we sought to determine if administering MSCs preconditioned with CpG in radiation-associated neutropenia would allow the host to better respond to systemic bacterial infection.
Methods
MSCs and CdM
Human bone marrow-derived MSCs were purchased from the Center for the Preparation and Distribution of Adult Stem Cells at Texas A&M University Health Science Center. The cells were characterized by Texas A&M and reported to meet all criteria for MSC phenotype established by the International Society for Cellular Therapy.21 We confirmed the MSC phenotype by flow cytometry analysis (see the “Results” section). Passage 5 MSCs were cultured to 80% to 90% confluency and conditioned using 3 µg/mL CpG-ODN 2336 (InvivoGen) for 30 minutes immediately before injection. Conditioned media (CdM) was prepared from passage 6 to 8 MSCs and incubated in supplement-free α MEM media (Gibco) for 24 hours. CdM was collected and concentrated using Amicon Ultra-15 centrifugal filter units with 3-KDa cutoff (Millipore) per the manufacturer’s instructions.
Experimental model
Eight- to 10-week-old male outbred CD-1 mice (Charles River) were used as previously described in pneumonia36-38 and sepsis39-41 models. Outbred mice were chosen because their genetic variability was thought to more closely reflect the genetic diversity of the general human population, particularly as polygenic traits can affect susceptibility to infections, for example with Pseudomonas, in both humans42 and mice.36 The mice were irradiated with 5 Gy radiation using a Gammacell 40 Exactor 137Cs dual-source irradiator (Best Theratronics). On day 3, animals were anesthetized with ketamine/xylazine and intracardiac injection with 200 µL of (1) phosphate-buffered saline (PBS) vehicle control, (2) 5 × 105 MSCs in PBS, or (3) 5 × 105 CpG-MSCs in PBS was performed. Intracardiac was chosen as the route of administration to better mimic the universality of central venous catheters for the administration of medications in the HSCT patient population.43 On day 7, mice were infected intranasally with Pseudomonas aeruginosa (ATCC 27853) in 40 µL PBS. Mice were monitored for 7 days for survival or euthanized 24 to 48 hours after infection for functional analyses. For survival studies, 6 to 7 × 106 colony-forming units (CFU) of P aeruginosa was used. For functional studies, 1 to 2 × 106 CFU of P aeruginosa was used to minimize censorship of deceased animals. Mice that died before the predetermined functional time point were not included in the analysis. In experiments using conditioned media, mice were injected with CdM from 2.5 × 105 MSCs in 100 µL on day 3 as described previously, and a second injection by tail vein with CdM from 2.5 × 105 MSCs in 100 µL was performed 6 hours after infection.
In vitro coculture of bone marrow-derived neutrophils with MSCs
MSCs (5000/well) were plated in a 96-well black clear bottom tissue culture plate (Thomas Scientific) and incubated overnight at 37°C with 5% CO2. The following day, CpG-MSCs were stimulated with CpG 2336 (3 µg/mL) for 30 minutes, washed with PBS and incubated in complete media for 24 hours. On day 3, mice that had received 5 Gy radiation 7 days prior were euthanized, and single cell suspensions of hematopoietic cells were prepared from bilateral femurs and tibias. Red cells were lysed with ammonium chloride and neutrophils were isolated by density centrifugation in Histopaque 1119 (Sigma) and Histopaque 1077 (Sigma) at 372g for 30 minutes at room temperature without brake. Neutrophils were collected at the interface, washed, and resuspended in Hanks balanced salt solution (HBSS; Gibco). 1 × 105 neutrophils were seeded into each well with P aeruginosa opsonized for 30 minutes with autologous serum, corresponding to 1 to 3 × 106 bacterial CFUs, and incubated for 2-3 hours at 37°C with 5% CO2.
Additional methods are available in the supplemental Methods.
Results
Characterization of human MSCs
A time course of fluorescein-CpG 2336 uptake by MSCs was performed to determine the optimal stimulation time (supplemental Figure 1A), which showed peak uptake of CpG by 30 minutes. Surface phenotyping was performed on MSCs with and without CpG stimulation per the minimal criteria outlined by the International Society for Cellular Therapy (supplemental Figure 1B) and demonstrated the anticipated absence of hematopoietic markers and the presence of mesenchymal markers. Expression of TLR9 was confirmed (supplemental Figure 1C) in MSCs, and CpG-MSCs, showing robust expression of TLR9 at baseline (41.8%) and after stimulation (54.1%). To confirm that CpG preconditioning protects MSCs from oxidative stress-induced death,35 cells were either untreated or preconditioned with CpG (3 µg/mL and 6 µg/mL) for 30 minutes before exposure to H2O2 (supplemental Figure 1D). In unconditioned MSCs, H2O2 significantly reduced cell viability by 50%. However, in MSCs preconditioned with CpG, cell death was significantly attenuated to the same degree with both 3 µg/mL and 6 µg/mL of CpG. Given these data, we used a preconditioning regimen of 3 µg/mL CpG for 30 minutes for the remainder of our experiments. Endotoxin testing was performed on reagents used in the preparation of MSCs (PBS, media) and whole cell lysates using the Limulus Amebocyte Lysate Chromogenic Endotoxin Assay Kit (Pierce) demonstrating <0.5 EU/mL.
Administration of CpG-MSCs after radiation-associated bone marrow hypoplasia confers a survival benefit when mice are infected with P aeruginosa
We first assessed the therapeutic impact of MSCs and CpG-MSCs in vivo (Figure 1A). In sublethally irradiated (5 Gy) mice that were subsequently challenged with P aeruginosa, mice treated with PBS had 40% mortality over 7 days (Figure 1B), which was significantly attenuated when mice were administered CpG-MSCs (10% mortality). This survival benefit was not observed when comparing mice that received unconditioned MSCs with mice that received PBS.
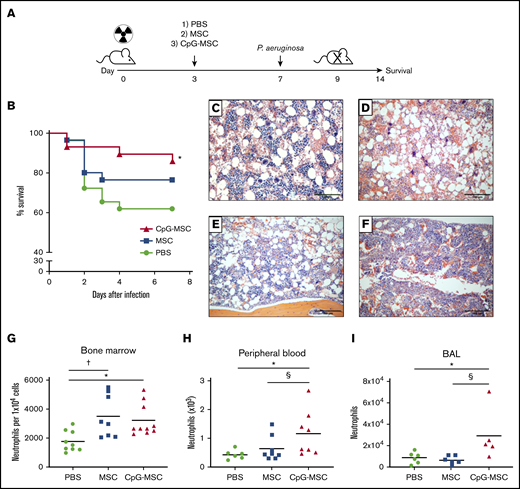
Improved survival and increased neutrophils in mice treated with CpG-MSCs in P aeruginosa pulmonary infection in the setting of radiation-associated bone marrow hypoplasia. (A) Schematic of experimental model. Mice were irradiated with 5 Gy on day 0. Three days later, mice received an injection of 1) PBS, 2) 5 × 105 MSCs, or 3) 5 × 105 CpG-MSCs. On day 7, the mice were infected intranasally with either 6 to 7 × 106 or 1 to 2 × 106 colony forming units of P aeruginosa and either monitored for 7 additional days for survival or euthanized 24 to 48 hours after infection for functional analyses, respectively. (B) Survival of male CD-1 mice (n = 30 per group) after irradiation with 5 Gy, injection with PBS (green filled circle), 5 × 105 MSC (blue filled square), or 5 × 105 MSC conditioned with CpG-MSC (red filled triangle) and infected with pulmonary P aeruginosa. Data are presented as Kaplan-Meier survival curves and analyzed by log-rank test. *Significant comparisons (CpG-MSC vs PBS, P = .048). Representative hematoxylin and eosin staining of femurs 9 days after irradiation with 5 Gy, (C) without infection vs (D-F) with P aeruginosa pulmonary infection and administration of (D) PBS, (E) 5 × 105 MSCs, or (F) 5 × 105 CpG-MSCs. Images were taken on a Nikon Eclipse 80i microscope using an AmScope 18MP USB 3.0 digital camera at 10× original magnification acquired on NIS-Elements BR 3.2 software and white-balanced in Fiji ImageJ. (G) Single cell suspensions were prepared from the bone marrow of irradiated mice administered PBS (n = 9, green filled circle), MSC (n = 8, blue filled square), or CpG-MSC (n = 10, red filled triangle) 48 hours after infection with P aeruginosa and cell populations were analyzed by cytometry by time of flight. Neutrophil numbers were assessed by analysis of variance (ANOVA), P = .007. *Significant comparisons by Bonferroni’s multiple comparisons test, PBS vs CpG-MSC, P = .029; †PBS vs MSC, P = .012. (H) Whole peripheral blood was collected 48 hours after infection in mice administered PBS (n = 6), MSC (n = 8), and CpG-MSC (n = 8), and total neutrophil and large unstained cell (immature granulocyte) counts were analyzed by Kruskal-Wallis, P = .025. *Significant comparisons by uncorrected Dunn’s post hoc test, PBS vs CpG-MSC, P = .016; §CpG-MSC vs MSC, P = .038. (I) Bronchoalveolar lavage (BAL) was obtained from mice administered PBS (n = 6), MSC (n = 6), or CpG-MSC (n = 5) 24 hours after infection with P aeruginosa and the number of neutrophils (GR-1 cells) analyzed by Kruskal-Wallis, P = .018. *Significant comparisons by Dunn’s post hoc test, PBS vs CpG-MSC, P = .044; §CpG-MSC vs MSC, P = .009.
Improved survival and increased neutrophils in mice treated with CpG-MSCs in P aeruginosa pulmonary infection in the setting of radiation-associated bone marrow hypoplasia. (A) Schematic of experimental model. Mice were irradiated with 5 Gy on day 0. Three days later, mice received an injection of 1) PBS, 2) 5 × 105 MSCs, or 3) 5 × 105 CpG-MSCs. On day 7, the mice were infected intranasally with either 6 to 7 × 106 or 1 to 2 × 106 colony forming units of P aeruginosa and either monitored for 7 additional days for survival or euthanized 24 to 48 hours after infection for functional analyses, respectively. (B) Survival of male CD-1 mice (n = 30 per group) after irradiation with 5 Gy, injection with PBS (green filled circle), 5 × 105 MSC (blue filled square), or 5 × 105 MSC conditioned with CpG-MSC (red filled triangle) and infected with pulmonary P aeruginosa. Data are presented as Kaplan-Meier survival curves and analyzed by log-rank test. *Significant comparisons (CpG-MSC vs PBS, P = .048). Representative hematoxylin and eosin staining of femurs 9 days after irradiation with 5 Gy, (C) without infection vs (D-F) with P aeruginosa pulmonary infection and administration of (D) PBS, (E) 5 × 105 MSCs, or (F) 5 × 105 CpG-MSCs. Images were taken on a Nikon Eclipse 80i microscope using an AmScope 18MP USB 3.0 digital camera at 10× original magnification acquired on NIS-Elements BR 3.2 software and white-balanced in Fiji ImageJ. (G) Single cell suspensions were prepared from the bone marrow of irradiated mice administered PBS (n = 9, green filled circle), MSC (n = 8, blue filled square), or CpG-MSC (n = 10, red filled triangle) 48 hours after infection with P aeruginosa and cell populations were analyzed by cytometry by time of flight. Neutrophil numbers were assessed by analysis of variance (ANOVA), P = .007. *Significant comparisons by Bonferroni’s multiple comparisons test, PBS vs CpG-MSC, P = .029; †PBS vs MSC, P = .012. (H) Whole peripheral blood was collected 48 hours after infection in mice administered PBS (n = 6), MSC (n = 8), and CpG-MSC (n = 8), and total neutrophil and large unstained cell (immature granulocyte) counts were analyzed by Kruskal-Wallis, P = .025. *Significant comparisons by uncorrected Dunn’s post hoc test, PBS vs CpG-MSC, P = .016; §CpG-MSC vs MSC, P = .038. (I) Bronchoalveolar lavage (BAL) was obtained from mice administered PBS (n = 6), MSC (n = 6), or CpG-MSC (n = 5) 24 hours after infection with P aeruginosa and the number of neutrophils (GR-1 cells) analyzed by Kruskal-Wallis, P = .018. *Significant comparisons by Dunn’s post hoc test, PBS vs CpG-MSC, P = .044; §CpG-MSC vs MSC, P = .009.
To identify sites of pathological difference between the 3 experimental groups, hematoxylin and eosin staining was performed on paraffin-embedded tissues harvested 48 hours after infection, and the most striking difference was observed in the bone marrow. Without infection, irradiated mice had moderately hypocellular bone marrow (Figure 1C). In the setting of P aeruginosa infection, there was a trend toward decreased cellularity (Figure 1D). However, this hypocellularity appeared to be attenuated when mice were given MSCs (Figure 1E) and CpG-MSCs (Figure 1F), although this was not statistically significant (supplemental Figure 2A). We then sought to determine if there were instead differences in cellular composition between the 3 groups, and found increased neutrophils in the bone marrow (Figure 1G) by cytometry by time of flight (CyTOF) in mice given MSCs and CpG-MSCs. Ly6G DAB staining was also performed in the bone marrow showing increased neutrophils in mice given CpG-MSCs (supplemental Figure 2B-E). There were no differences in the numbers of other cell populations, including macrophages, T cells, and B cells (supplemental Figure 2F-H). Despite increased numbers of Ly6G+ neutrophils in mice given both MSCs and CpG-MSCs, it was only in the mice given CpG-MSCs that an increased number of neutrophils were found in the peripheral blood (Figure 1H) and at the site of infection (lung; Figure 1I).
To further characterize the cell population(s) present within the bone marrow, we used CyTOF technology. Single cell suspensions were prepared from the bone marrow of irradiated mice in our 3 experimental conditions and labeled with 33 immunophenotyping markers (supplemental Table 1). Dimensionality reduction was performed using t-SNE, and manual gating was performed to identify general cluster phenotypes (Figure 2A). Using X-shift algorithm for K-nearest neighbor estimation, the optimal number of clusters was determined by the elbow point of k = 60, which corresponded to 44 clusters. We rounded the number up to 45 and ran a SPADE algorithm to identify 45 clusters of phenotypically similar cells (Figure 2B). Post hoc analysis identified 2 neutrophil clusters (Figure 2C), cluster 23 (green) and cluster 28 (red), that were significantly increased in the mice that received CpG-MSCs compared with PBS, with a trend toward an increase when comparing mice that received MSCs with mice that received PBS (Figure 2D). A heatmap of the log transformed mean expression levels of the immunophenotyping markers in each cluster was generated (Figure 2E; for full table, see supplemental Figure 3), demonstrating that the most significant cluster, cluster 28, comprised Ly6GlowKi-67+PU.1+ cells, suggesting that proliferating neutrophils were increased within the bone marrow. Notably, cluster 23 comprises a population of CXCR2+-expressing neutrophils, which have been shown to be involved in neutrophil recruitment.44 This suggests that CpG-MSCs may also contribute to neutrophil egress from the bone marrow into the periphery and to the site of infection. To demonstrate that the cells themselves were not promoting a granulopoiesis response without the presence of infection, we gave mice PBS, MSCs, or CpG-MSCs 3 days after radiation and harvested the bone marrow 8 days after radiation for quantification of myeloid cell populations (supplemental Figure 4A). CyTOF analysis demonstrated no significant difference in neutrophil or macrophage numbers among the 3 groups (supplemental Figure 4B-C).
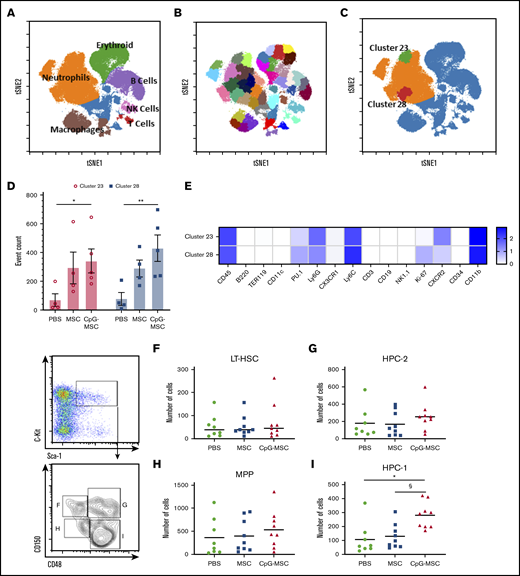
Cytometry by time of flight identifies 2 neutrophil clusters that are increased in mice that received CpG-MSCs compared with mice that received PBS; flow cytometry further identifies a CD150−CD48+ restricted progenitor population that is expanded in mice that received CpG-MSC. Single cell suspensions from the bone marrow of irradiated mice that received PBS (n = 4), MSC (n = 5), and CpG-MSC (n = 5) prepared 48 hours after infection with P aeruginosa were labeled with 33 immunophenotyping markers. (A) A tSNE plot of concatenated data from PBS, MSC, and CpG-MSC groups after equal sampling (10 000 events per file). Manual gating identified distinct clusters of Ly6G+ neutrophils (orange), TER-119+ erythroid cells (green), CD19+ B cells (purple), CD3+ T cells (red), NK cells (pink), and F4/80+ macrophages (brown). Cells not otherwise identified by immunophenotyping markers are represented in blue. (B) Forty-five clusters were identified using SPADE algorithm and overlaid on a tSNE plot of concatenated data. (C) Data were assessed by 2-way ANOVA, interaction P = .0002. 2 clusters, cluster 28 (red) and cluster 23 (green) were further identified as significantly different between PBS and CpG-MSC groups, but not PBS vs MSC, by post hoc multiple comparisons testing. (D) Event counts of the number of cells in cluster 23 (○, pink bars) and cluster 28 (▪, blue bars) in mice that received PBS, MSC, and CpG-MSC are shown. Post hoc analysis by Dunnett’s multiple comparison test was performed. *Significant comparisons, PBS vs CpG-MSC, cluster 23, P = .029); **PBS vs CpG-MSC, cluster 28, P = .003. (E) Log-transformed mean expression levels of select immunophenotyping markers (columns) by cluster number (rows) in clusters 23 and 28 from data concatenated from irradiated and infected mice given PBS (n = 4), MSC (n = 5), and CpG-MSC (n = 5). Flow cytometric analysis of primitive progenitors demonstrate no significant difference between mice given PBS (n = 8, green filled circle), MSC (n = 9, blue filled square), and CpG-MSC (n = 9, red filled triangle) by ANOVA in the numbers of (F) long-term hematopoietic stem cells (LT-HSC; Lin−Sca1+C-kit+CD150+CD48−) or (H) multipotent progenitors (MPP; Lin−Sca1+C-kit+CD150−CD48−) cells. (I) There was an increased number of Lin−Sca1+C-kit+CD150−CD48+ restricted progenitor cells (HPC-1) but not (G) Lin−Sca1+C-kit+CD150+CD48+ restricted progenitor cells (HPC-2) in mice that received CpG-MSC. Data were analyzed by ANOVA, P = .002. *Significant comparisons by Bonferroni’s multiple comparisons test, PBS vs CpG-MSC, P = .005; §CpG-MSC vs MSC, P = .010. tSNE, t-distributed stochastic neighbor embedding.
Cytometry by time of flight identifies 2 neutrophil clusters that are increased in mice that received CpG-MSCs compared with mice that received PBS; flow cytometry further identifies a CD150−CD48+ restricted progenitor population that is expanded in mice that received CpG-MSC. Single cell suspensions from the bone marrow of irradiated mice that received PBS (n = 4), MSC (n = 5), and CpG-MSC (n = 5) prepared 48 hours after infection with P aeruginosa were labeled with 33 immunophenotyping markers. (A) A tSNE plot of concatenated data from PBS, MSC, and CpG-MSC groups after equal sampling (10 000 events per file). Manual gating identified distinct clusters of Ly6G+ neutrophils (orange), TER-119+ erythroid cells (green), CD19+ B cells (purple), CD3+ T cells (red), NK cells (pink), and F4/80+ macrophages (brown). Cells not otherwise identified by immunophenotyping markers are represented in blue. (B) Forty-five clusters were identified using SPADE algorithm and overlaid on a tSNE plot of concatenated data. (C) Data were assessed by 2-way ANOVA, interaction P = .0002. 2 clusters, cluster 28 (red) and cluster 23 (green) were further identified as significantly different between PBS and CpG-MSC groups, but not PBS vs MSC, by post hoc multiple comparisons testing. (D) Event counts of the number of cells in cluster 23 (○, pink bars) and cluster 28 (▪, blue bars) in mice that received PBS, MSC, and CpG-MSC are shown. Post hoc analysis by Dunnett’s multiple comparison test was performed. *Significant comparisons, PBS vs CpG-MSC, cluster 23, P = .029); **PBS vs CpG-MSC, cluster 28, P = .003. (E) Log-transformed mean expression levels of select immunophenotyping markers (columns) by cluster number (rows) in clusters 23 and 28 from data concatenated from irradiated and infected mice given PBS (n = 4), MSC (n = 5), and CpG-MSC (n = 5). Flow cytometric analysis of primitive progenitors demonstrate no significant difference between mice given PBS (n = 8, green filled circle), MSC (n = 9, blue filled square), and CpG-MSC (n = 9, red filled triangle) by ANOVA in the numbers of (F) long-term hematopoietic stem cells (LT-HSC; Lin−Sca1+C-kit+CD150+CD48−) or (H) multipotent progenitors (MPP; Lin−Sca1+C-kit+CD150−CD48−) cells. (I) There was an increased number of Lin−Sca1+C-kit+CD150−CD48+ restricted progenitor cells (HPC-1) but not (G) Lin−Sca1+C-kit+CD150+CD48+ restricted progenitor cells (HPC-2) in mice that received CpG-MSC. Data were analyzed by ANOVA, P = .002. *Significant comparisons by Bonferroni’s multiple comparisons test, PBS vs CpG-MSC, P = .005; §CpG-MSC vs MSC, P = .010. tSNE, t-distributed stochastic neighbor embedding.
Mice that receive CpG-MSCs have greater myeloid differentiation and proliferation potential in the setting of P aeruginosa infection, an effect that is TLR9 dependent
To further validate the CyTOF data, we performed flow cytometry to identify hematopoietic progenitor populations that could explain the increase in neutrophils. There were no differences in the number of Lin−Sca1+C-kit+ cells (supplemental Figure 5A), LT-HSC (Figure 2F), hematopoietic progenitor cells (HPC-2; Figure 2G), or MPP(Figure 2H). We found 1 population of lineage-restricted progenitors (Lin−Sca1+C-kit+CD150−CD48+; HPC-1) was expanded in mice that received CpG-MSCs compared with MSCs and PBS (Figure 2I). Notably, this expansion was not at the expense of more primitive long-term HSC, which have the greatest self-renewing capacity and contributes to marrow repopulation. These data suggest that mice given CpG-MSCs have greater progenitor cell proliferation and neutrophil differentiation compared with mice given MSCs alone or PBS.
To study the capacity of c-kit+ hematopoietic stem cells to proliferate and differentiate into myeloid cells, colony-forming assays were performed on c-kit+ cells from the bone marrow of mice in the experimental conditions. Representative colonies identified phenotypically were isolated after growth and analyzed by flow cytometry to confirm the presence of Ly6G+ neutrophils in granulocyte CFUs (CFU-G; Figure 3A), F4/80+ macrophages in macrophage CFUs (CFU-M; Figure 3B), and the presence of both cell types in granulocyte-macrophage CFUs (CFU-GM; Figure 3C). Mice that received MSCs had a twofold greater myeloid differentiation potential compared with mice that received PBS, an effect that is further augmented when the MSCs are preconditioned with CpG (Figure 3D). Notably, when mice were challenged with P aeruginosa 24 hours after injection with MSCs or CpG-MSCs (supplemental Figure 5B), an increase in myeloid proliferation and differentiation potential was not observed compared with PBS control (supplemental Figure 5C). This suggests that it can take up to 72 hours for the in vivo expansion of HPC-1 progenitor cells in CpG-MSC-treated animals to occur, representing a lag before prophylactic efficacy may be observed at this given level of radiation injury
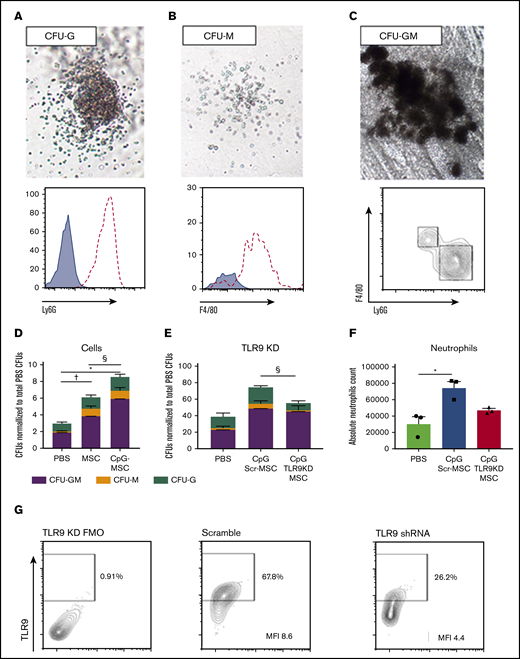
Irradiated mice given CpG-MSCs have greater myeloid differentiation and proliferation potential after infection compared with mice given MSC and PBS alone, an effect that is partially TLR9 dependent. Representative images of (A) CFU-G, (B) CFU-M, and (C) CFU-GM. Images were taken on a VWR Trinocular Inverted Microscope using an OMAX 18.0MP USB 3.0 digital camera at 4× original magnification acquired on ToupView software and cropped in Fiji ImageJ. Representative colonies were selected to confirm the presence of Ly6G+ neutrophils (dashed line) in CFU-Gs, F4/80+ macrophages (dashed line) in CFU-Ms compared to unstained controls (blue filled curve), and both Ly6G+ neutrophils and F4/80+ macrophages in CFU-GMs. (D) 1000 c-kit+ cells isolated from the bone marrow of irradiated mice administered PBS, MSC, or CpG-MSC (n = 3 per group) and infected with pulmonary P aeruginosa were seeded into methylcellulose media in triplicate. Cultures were grown for 9 days before blinded manual determination of CFU-GM (purple bars), CFU-M (orange bars), or CFU-G (green bars). Data were assessed by 2-way ANOVA; interaction P = .005. †Significant comparisons of CFU-GMs by Bonferroni’s multiple comparisons test, PBS vs MSC, P = .012; *PBS vs CpG-MSC, P < .0001; and §CpG-MSC vs MSC, P = .011. (E) CFU counts when irradiated and infected mice were given PBS, CpG-MSCs, or CpG TLR9KD-MSCs (n = 3 per group). Data were assessed by 2-way ANOVA; interaction P < .0001. §Significant comparison of CFU-Gs by Bonferroni’s multiple comparisons test, CpG Scr-MSC vs CpG TLR9KD MSC, P = .011. (F) Quantification of the total neutrophil count per plate after 9 days of growth from 1000 c-kit+ cells harvested from mice that received PBS (green bars), CpG Scr-MSCs (blue bars), or CpG TLR9KD-MSCs (red bars). Data were assessed by 2-way ANOVA; interaction P = .009. *Significant comparisons by Bonferroni’s multiple comparisons test, PBS vs CpG-MSCs, P = .010. (G) TLR9 protein expression by flow cytometry in fluorescence minus 1 (FMO) wild-type control, MSCs transduced with scramble shRNA (MFI 8.6) and MSCs transduced with TLR9 shRNA (MFI 4.4). Based on differences in MFI, there is ∼50% TLR9 protein knockdown in TLR9KD MSCs.
Irradiated mice given CpG-MSCs have greater myeloid differentiation and proliferation potential after infection compared with mice given MSC and PBS alone, an effect that is partially TLR9 dependent. Representative images of (A) CFU-G, (B) CFU-M, and (C) CFU-GM. Images were taken on a VWR Trinocular Inverted Microscope using an OMAX 18.0MP USB 3.0 digital camera at 4× original magnification acquired on ToupView software and cropped in Fiji ImageJ. Representative colonies were selected to confirm the presence of Ly6G+ neutrophils (dashed line) in CFU-Gs, F4/80+ macrophages (dashed line) in CFU-Ms compared to unstained controls (blue filled curve), and both Ly6G+ neutrophils and F4/80+ macrophages in CFU-GMs. (D) 1000 c-kit+ cells isolated from the bone marrow of irradiated mice administered PBS, MSC, or CpG-MSC (n = 3 per group) and infected with pulmonary P aeruginosa were seeded into methylcellulose media in triplicate. Cultures were grown for 9 days before blinded manual determination of CFU-GM (purple bars), CFU-M (orange bars), or CFU-G (green bars). Data were assessed by 2-way ANOVA; interaction P = .005. †Significant comparisons of CFU-GMs by Bonferroni’s multiple comparisons test, PBS vs MSC, P = .012; *PBS vs CpG-MSC, P < .0001; and §CpG-MSC vs MSC, P = .011. (E) CFU counts when irradiated and infected mice were given PBS, CpG-MSCs, or CpG TLR9KD-MSCs (n = 3 per group). Data were assessed by 2-way ANOVA; interaction P < .0001. §Significant comparison of CFU-Gs by Bonferroni’s multiple comparisons test, CpG Scr-MSC vs CpG TLR9KD MSC, P = .011. (F) Quantification of the total neutrophil count per plate after 9 days of growth from 1000 c-kit+ cells harvested from mice that received PBS (green bars), CpG Scr-MSCs (blue bars), or CpG TLR9KD-MSCs (red bars). Data were assessed by 2-way ANOVA; interaction P = .009. *Significant comparisons by Bonferroni’s multiple comparisons test, PBS vs CpG-MSCs, P = .010. (G) TLR9 protein expression by flow cytometry in fluorescence minus 1 (FMO) wild-type control, MSCs transduced with scramble shRNA (MFI 8.6) and MSCs transduced with TLR9 shRNA (MFI 4.4). Based on differences in MFI, there is ∼50% TLR9 protein knockdown in TLR9KD MSCs.
To study the dependence of CpG preconditioning on the TLR9 pathway in MSCs, we transduced MSCs with TLR9 short hairpin RNA (shRNA) in a lentiviral vector. Using this method, we achieved approximately 50% knock down of TLR9 protein expression in MSCs (TLR9KD-MSC; Figure 3G) by mean fluorescence intensity (MFI, 4.4) compared with MSCs transduced with scramble shRNA (Scr-MSC, MFI 8.6). Irradiated mice were then injected with PBS control, Scr-MSCs preconditioned with CpG, or TLR9KD-MSCs preconditioned with CpG prior to P aeruginosa infection. The c-kit+ cells purified from mice that received CpG Scr-MSCs demonstrated a twofold increase in total colony formation as wild-type CpG-MSCs compared with c-kit+ cells from mice that received PBS (Figure 3E). Conversely, mice that received CpG TLR9KD-MSCs demonstrated a lower (1.4 times) increase in colony count compared with mice that received PBS. Notably, there was a statistically significant difference in the number of CFU-G between mice that received CpG Scr-MSCs and mice that received CpG TLR9KD-MSCs (P = .0110). This was further confirmed by quantifying the total neutrophil count per plate after colony growth, which demonstrated increased neutrophils in mice that received CpG Scr-MSCs compared with PBS, but not in mice that received CpG TLR9KD-MSCs (Figure 3F). Overall, these data suggest that treating neutropenic mice with CpG-MSCs results in augmented granulopoiesis in response to infection, an effect that is partially dependent on TLR9.
Neutrophils exposed to CpG-MSCs produce fewer neutrophil extracellular traps, which was associated with decreased organ damage
Despite increased neutrophils in mice given CpG-MSCs, assessment of organ bacterial CFUs in the left whole lung 48 hours after infection did not demonstrate a reduction in bacterial clearance among the 3 groups (Figure 4A). Instead, mice given CpG-MSCs had decreased organ damage, as assessed by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining as a marker for cellular apoptosis and death. TUNEL staining of kidney (Figure 4B) and left lung (Figure 4C) showed less TUNEL-positive staining in mice that received CpG-MSCs, compared with mice that received PBS. There was a similar trend that did not meet statistical significance in the spleen, and there was no difference in the liver (supplemental Figure 6A-B). Representative whole tissue sections can be found in supplemental Figure 6C-H. This decreased organ damage also corresponded with lower levels of systemic interleukin (IL)-6 in the plasma of mice that received CpG-MSCs (Figure 4D), suggesting that these mice had less severe inflammation than the mice that did not receive MSCs. Taken together, these data suggest that the neutrophils expanded in vivo by CpG-MSCs in response to infection were less organ destructive.
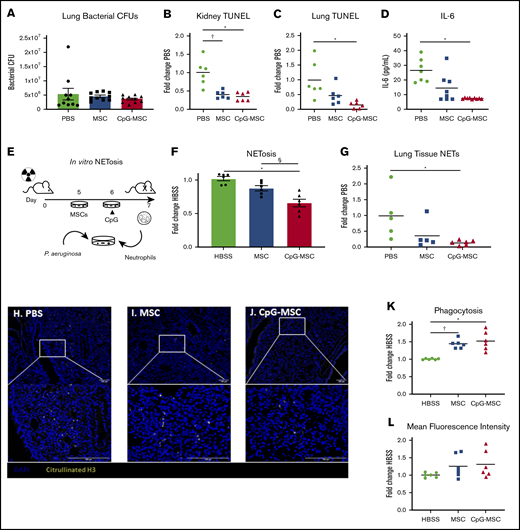
Irradiated mice that received CpG-MSCs have less organ damage after infection. (A) Irradiated mice given PBS, MSCs, or CpG-MSCs were euthanized 48 hours after P aeruginosa infection and the whole left lung was harvested for homogenization and quantification of organ bacterial CFU. There was no significant difference by Kruskal-Wallis in bacterial clearance between mice that received PBS (green bars), MSC (blue bars), or CpG-MSC (red bars) (n = 10 per group). TUNEL staining of (B) kidney (n = 6 per group) and (C) left lung (n = 6 per group), standardized to total tissue area, relative to TUNEL staining in mice that received PBS (fold change PBS). Organs were harvested 48 hours after P aeruginosa infection in mice that received PBS (green filled circle), MSC (blue filled square), or CpG-MSC (red filled triangle). Data were assessed by ANOVA, P = .0002 (kidneys), P = .009 (lung). *Significant comparisons by Bonferroni’s multiple comparisons test, PBS vs CpG-MSC, P = .0005 (kidneys), P = .008 (lungs); †PBS vs MSC, P = .001 (kidneys). (D) IL-6 Luminex on plasma samples (n = 7 PBS, n = 8 MSC, n = 9 CpG-MSC) obtained 48 hours after infection analyzed by Kruskal-Wallis, P = .0012. *Significant comparisons by Dunn’s multiple comparisons test, PBS vs CpG, P = .001. (E) Experimental design for the in vitro analysis of NET formation. Mice are irradiated with 5 Gy radiation on day 0. Two days before the assay, MSCs are plated. The following day, CpG-MSCs are stimulated with CpG (3 µg/mL) for 30 minutes. On the day of the assay, bone marrow neutrophils are harvested from irradiated mice by density centrifugation and added to MSCs, CpG-MSCs or media control with opsonized P aeruginosa. (F) Bone marrow-derived neutrophils from irradiated mice were cocultured with HBSS, MSC, or CpG-MSCs and P aeruginosa for 3 hours before assay by Sytox Green. Data were analyzed by pooled 1-way ANOVA (n = 6 per group), P = .0001. *Significant comparisons by Bonferroni’s multiple comparisons test, CpG-MSC vs HBSS, P = .0001; §CpG-MSC vs MSC, P = .009. Immunofluorescence for citrullinated H3 (yellow) was performed on lung tissue sections from irradiated and infected mice given (H) PBS, (I) MSCs, or (J) CpG-MSCs demonstrating (G) decreased citrullinated H3 staining in the lungs of mice given CpG-MSCs. Images were taken on a Nikon Eclipse 80i using an AmScope 18MP USB 3.0 digital camera at 20× and 40× original magnification acquired on NIS-Elements BR 3.2 software with DAPI and AlexaFluor 488 fluorochromes in Vectashield antifade mounting media (Vector); background was subtracted, and brightness/contrast were adjusted to whole channel images before merging the channels in Fiji ImageJ. Coculturing bone marrow-derived neutrophils from irradiated mice with MSCs and CpG-MSCs resulted in (K) more neutrophils phagocytosing at least 1 P aeruginosa. Data were analyzed by Brown-Forsythe ANOVA, P = .003. †Significant comparisons by Dunnett’s multiple comparisons test, PBS vs MSC, P = .001; *PBS vs CpG-MSC, P = .015. There was no significant difference in the (L) mean fluorescence intensity of phagocytosed P aeruginosa.
Irradiated mice that received CpG-MSCs have less organ damage after infection. (A) Irradiated mice given PBS, MSCs, or CpG-MSCs were euthanized 48 hours after P aeruginosa infection and the whole left lung was harvested for homogenization and quantification of organ bacterial CFU. There was no significant difference by Kruskal-Wallis in bacterial clearance between mice that received PBS (green bars), MSC (blue bars), or CpG-MSC (red bars) (n = 10 per group). TUNEL staining of (B) kidney (n = 6 per group) and (C) left lung (n = 6 per group), standardized to total tissue area, relative to TUNEL staining in mice that received PBS (fold change PBS). Organs were harvested 48 hours after P aeruginosa infection in mice that received PBS (green filled circle), MSC (blue filled square), or CpG-MSC (red filled triangle). Data were assessed by ANOVA, P = .0002 (kidneys), P = .009 (lung). *Significant comparisons by Bonferroni’s multiple comparisons test, PBS vs CpG-MSC, P = .0005 (kidneys), P = .008 (lungs); †PBS vs MSC, P = .001 (kidneys). (D) IL-6 Luminex on plasma samples (n = 7 PBS, n = 8 MSC, n = 9 CpG-MSC) obtained 48 hours after infection analyzed by Kruskal-Wallis, P = .0012. *Significant comparisons by Dunn’s multiple comparisons test, PBS vs CpG, P = .001. (E) Experimental design for the in vitro analysis of NET formation. Mice are irradiated with 5 Gy radiation on day 0. Two days before the assay, MSCs are plated. The following day, CpG-MSCs are stimulated with CpG (3 µg/mL) for 30 minutes. On the day of the assay, bone marrow neutrophils are harvested from irradiated mice by density centrifugation and added to MSCs, CpG-MSCs or media control with opsonized P aeruginosa. (F) Bone marrow-derived neutrophils from irradiated mice were cocultured with HBSS, MSC, or CpG-MSCs and P aeruginosa for 3 hours before assay by Sytox Green. Data were analyzed by pooled 1-way ANOVA (n = 6 per group), P = .0001. *Significant comparisons by Bonferroni’s multiple comparisons test, CpG-MSC vs HBSS, P = .0001; §CpG-MSC vs MSC, P = .009. Immunofluorescence for citrullinated H3 (yellow) was performed on lung tissue sections from irradiated and infected mice given (H) PBS, (I) MSCs, or (J) CpG-MSCs demonstrating (G) decreased citrullinated H3 staining in the lungs of mice given CpG-MSCs. Images were taken on a Nikon Eclipse 80i using an AmScope 18MP USB 3.0 digital camera at 20× and 40× original magnification acquired on NIS-Elements BR 3.2 software with DAPI and AlexaFluor 488 fluorochromes in Vectashield antifade mounting media (Vector); background was subtracted, and brightness/contrast were adjusted to whole channel images before merging the channels in Fiji ImageJ. Coculturing bone marrow-derived neutrophils from irradiated mice with MSCs and CpG-MSCs resulted in (K) more neutrophils phagocytosing at least 1 P aeruginosa. Data were analyzed by Brown-Forsythe ANOVA, P = .003. †Significant comparisons by Dunnett’s multiple comparisons test, PBS vs MSC, P = .001; *PBS vs CpG-MSC, P = .015. There was no significant difference in the (L) mean fluorescence intensity of phagocytosed P aeruginosa.
To test this hypothesis, we next assessed neutrophil function. Given evidence that neutrophil extracellular traps (NETs) contribute to mortality in a murine model of endotoxemic shock,45 we sought to determine if CpG-MSCs could inhibit NETosis. To study this in vitro, we used a coculture assay and visualization of extracellular nucleic acid using Sytox green, a cell membrane impermeable stain (Figure 4E). Using this assay, neutrophils cocultured with CpG-MSCs had significantly less NET formation than when neutrophils were cocultured with MSCs alone or HBSS control (Figure 4F). We confirmed this finding in vivo with immunofluorescence staining and quantification of citrullinated H3 in the lungs (Figure 4G), demonstrating less citrullinated H3-positive cells in mice that received CpG-MSCs (Figure 4H-J).
Enhanced neutrophil phagocytosis and bacterial clearance in the presence of MSCs in other murine models of sepsis in immunocompetent animals has been described.23,46 In line with this, there was a greater percentage of bone marrow-derived neutrophils phagocytosing at least 1 P aeruginosa bacterium when cocultured with MSCs and CpG-MSCs (Figure 4K) compared with HBSS controls. However, there was no difference in the mean fluorescence intensity, or average number of bacteria phagocytosed per neutrophil, among the 3 groups (Figure 4L). Representative flow cytometry plots can be found in supplemental Figure 6I. Taken together, CpG-MSCs expand neutrophils in vivo and more neutrophils participate in phagocytosis in cocultures with MSCs. However, these processes are balanced by decreased NET formation, which can normally help clear bacteria.47 Thus, in our model, CpG-MSCs strike a fine balance between inflammation and bacterial clearance that tips the scale toward survival in neutropenic sepsis.
CpG-MSCs exert their effects in the neutropenic host primarily by paracrine actions
MSCs are known to secrete a large repertoire of bioactive molecules, which is thought to be a major mechanism of therapeutic action.26,48 Given this, we sought to determine if MSCs were exerting their effect by paracrine actions, and if those actions could be augmented by preconditioning MSCs with CpG. To answer these questions, a modified experimental protocol using only conditioned media was used (Figure 5A). A second dose of CdM/control was given because viable MSCs were found in the bone marrow 3 days after injection (Figure 5B) that can continue to exert paracrine actions at the time of infection. Interestingly, c-kit+ cells from mice given CpG-MSC CdM formed more myeloid colonies compared with mice given CdM from unconditioned MSCs or media control (Figure 5C). Notably, the fold change increase (twofold) in total CFUs in mice given CpG-MSC CdM compared with control was similar to the fold change increase observed when mice were given CpG-MSC cells compared with control. This suggests that the effect of MSCs in the bone marrow is primarily driven by paracrine factors, and that preconditioning MSCs with CpG can alter the secretome to further augment its effects on granulopoiesis. To identify a soluble factor secreted by CpG-conditioned MSCs that could be driving the difference between MSCs and CpG-MSCs, we performed multiplexed cytokine analysis of the conditioned media comparing the 2 groups. Although both MSCs and CpG-MSCs secrete IL-6 and granulocyte colony-stimulating factor (G-CSF) that could be contributing to augmented granulopoiesis, there was no significant difference in the amount secreted by the 2 groups (Figure 5D). Other important factors implicated in emergency granulopoiesis including tumor necrosis factor-α, IL-1β, and FLT-3 ligand were secreted in very low (<10 pg/mL) amounts. Notably, both MSCs and CpG-MSCs secreted very high amounts of platelet-derived growth factor-AA (PDGF-AA), with more PDGF-AA secreted after CpG preconditioning. Previously published in vitro has demonstrated that PDGF can stimulate CFU-GM colony formation in a dose-dependent manner, which may explain the augmented granulopoiesis observed in this study.49
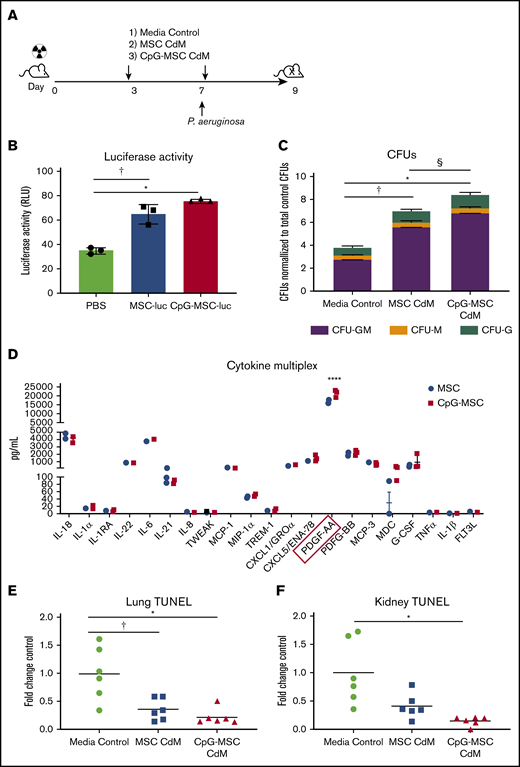
Increased myelopoiesis in response to infection is mediated by paracrine factors. (A) Schema of conditioned media experiments. Mice were irradiated (5 Gy) on day 0, followed by injection of 100 µL of CdM (dashed arrows) or media control on day 3. Mice were intranasally infected with P aeruginosa on day 7 and given a second dose of 100 µL CdM by tail vein 6 hours after infection. Mice were euthanized 9 days after irradiation for functional analysis. (B) Luciferase activity from 2 × 106 lysed bone marrow cells harvested from mice given PBS, MSC-luciferase (MSC-luc), and CpG-MSC-luciferase (CpG-MSC-luc) after 5 Gy radiation. Data were analyzed by ANOVA, P = .0001. †Significant comparisons by Bonferroni’s multiple comparisons test, PBS vs MSC, P = .0009; *PBS vs CpG-MSC, P = .0002. (C) A total of 1000 c-kit+ cells isolated from the bone marrow of irradiated mice given CdM prepared from MSCs or CpG-MSCs or media control (n = 3 per group per experiment) and infected with pulmonary P aeruginosa were seeded into methylcellulose media in triplicate and total colony count quantified. Data were assessed by 2-way ANOVA; interaction P < .0001. †Significant comparisons by Bonferroni’s multiple comparisons test, PBS vs MSC, P < .0001; *PBS vs CpG-MSC, P < .0001; §CpG-MSC vs MSC, P < .0001. (D) Multiplex cytokine analysis of CdM from MSCs and CpG-MSCs (n = 3 per group). Data were analyzed by 2-way ANOVA; interaction P < .0001. ****Significant comparison by Bonferroni’s multiple comparisons test, PDGF-AA, MSC vs CpG-MSC, P < .0001. TUNEL staining of (E) left lung (n = 6 per group) and (F) kidney (n = 6 per group), standardized to total tissue area, relative to TUNEL staining in mice that received media control (fold change control). Data were assessed by Brown-Forsythe ANOVA, P = .013 (kidney), P = .006 (lungs). *Significant comparisons by Dunnett’s multiple comparisons test, control vs CpG-MSC, P = .023 (lung), P = .040 (kidney); †control vs MSC, P = .049 (lung).
Increased myelopoiesis in response to infection is mediated by paracrine factors. (A) Schema of conditioned media experiments. Mice were irradiated (5 Gy) on day 0, followed by injection of 100 µL of CdM (dashed arrows) or media control on day 3. Mice were intranasally infected with P aeruginosa on day 7 and given a second dose of 100 µL CdM by tail vein 6 hours after infection. Mice were euthanized 9 days after irradiation for functional analysis. (B) Luciferase activity from 2 × 106 lysed bone marrow cells harvested from mice given PBS, MSC-luciferase (MSC-luc), and CpG-MSC-luciferase (CpG-MSC-luc) after 5 Gy radiation. Data were analyzed by ANOVA, P = .0001. †Significant comparisons by Bonferroni’s multiple comparisons test, PBS vs MSC, P = .0009; *PBS vs CpG-MSC, P = .0002. (C) A total of 1000 c-kit+ cells isolated from the bone marrow of irradiated mice given CdM prepared from MSCs or CpG-MSCs or media control (n = 3 per group per experiment) and infected with pulmonary P aeruginosa were seeded into methylcellulose media in triplicate and total colony count quantified. Data were assessed by 2-way ANOVA; interaction P < .0001. †Significant comparisons by Bonferroni’s multiple comparisons test, PBS vs MSC, P < .0001; *PBS vs CpG-MSC, P < .0001; §CpG-MSC vs MSC, P < .0001. (D) Multiplex cytokine analysis of CdM from MSCs and CpG-MSCs (n = 3 per group). Data were analyzed by 2-way ANOVA; interaction P < .0001. ****Significant comparison by Bonferroni’s multiple comparisons test, PDGF-AA, MSC vs CpG-MSC, P < .0001. TUNEL staining of (E) left lung (n = 6 per group) and (F) kidney (n = 6 per group), standardized to total tissue area, relative to TUNEL staining in mice that received media control (fold change control). Data were assessed by Brown-Forsythe ANOVA, P = .013 (kidney), P = .006 (lungs). *Significant comparisons by Dunnett’s multiple comparisons test, control vs CpG-MSC, P = .023 (lung), P = .040 (kidney); †control vs MSC, P = .049 (lung).
Given that organ damage is known to be an independent risk factor for mortality in sepsis,50 we wanted to know if CdM from CpG-MSCs also reduces damage to this important parameter. TUNEL staining of lung sections from irradiated and infected mice showed less TUNEL-positive staining in mice that received CdM from both CpG-conditioned and unconditioned MSCs compared with mice that received media control (Figure 5E). In the kidneys, only mice that received CdM from CpG-MSCs had less damage compared with media control (Figure 5F).
Discussion
In this paper, we demonstrated that MSCs given prophylactically to mice with radiation-associated neutropenia can enhance emergency granulopoiesis in response to P aeruginosa pneumo-sepsis, an effect that is further enhanced when MSCs are preconditioned with CpG.
“Emergency” or “demand adapted” granulopoiesis is mechanistically distinct from steady-state granulopoiesis.51,52 Important insights into this fundamental process have demonstrated that G-CSF is a critical regulator of neutrophil mobilization and response53 and that endothelial cells are the main catalysts for LPS-induced G-CSF-mediated switch to emergency granulopoiesis.16 However, in the presence of live pathogens in either immunocompetent16 or G-CSF−/−/GM-CSF−/− mice,54,55 redundant pathways appear to exist to mediate emergency granulopoiesis. Examples of alternative pathways include IL-6/IL-6R complex in LPS challenged G-CSF−/−/GM-CSF−/− mice54 and C-X-C motif chemokine ligand 1 in Streptococcus pneumoniae-associated pneumo-sepsis.56 Both G-CSF and IL-6 are well-established myelopoiesis factors; however, higher levels of IL-6 and G-CSF have been associated with worse clinical outcomes in sepsis.57,58
In this paper, we sought to harness the dual effects of MSCs as mediators of hematopoiesis and peripheral anti-inflammatory agents59,60 in a single therapeutic candidate, and to dissect the mechanisms of efficacy. We first demonstrated that administration of CpG-MSCs to neutropenic mice conferred a survival benefit when challenged with P aeruginosa pneumo-sepsis compared with PBS control. There has been immense interest in the use of MSCs both for the ex vivo expansion of HSC,61 as well as for the treatment of sepsis.46,62 Combining these 2 potential therapeutic aspects makes MSC-based cellular therapy an attractive candidate for the treatment of neutropenic sepsis. To our knowledge, there has only been 1 description of the use of MSCs in neutropenic sepsis,63 but the data remain unpublished. Our current study lends mechanistic insight into this therapeutic potential.
It has been demonstrated that bone marrow MSCs and related C-X-C motif chemokine ligand 12-abundant reticular cells can respond to circulating TLR ligands to regulate monocytic egress from the bone marrow in response to Listeria.22 However, MSCs have not definitively been shown to contribute to a demand-adapted neutrophil response in vivo before this study. Here, we demonstrate that exogenously administered MSCs can increase myeloid progenitor cell proliferation and differentiation in response to infection primarily through paracrine factors, an effect that is further enhanced when MSCs are preconditioned with CpG, a TLR9 agonist. We additionally found that when mice were infected with P aeruginosa 1 day after injection of MSCs or CpG-MSCs, an increase in myeloid proliferation and differentiation potential was not observed. We hypothesize that this lag in prophylactic efficacy is dependent on the expansion of HPC-1 progenitor cells, which in turn is dependent on the extent of damage to the hematopoietic niche from radiation injury64,65 and the size of the pool of hematopoietic progenitor cells that exist to repopulate the bone marrow. Undoubtedly, the complex interplay between radiation damage (lethal vs sublethal), stem cell renewal and repopulation (in the context of transplantation vs radiation injury alone), and the ability of MSCs to modify these variables, warrants further investigation to better delineate the kinetics of this effect.
In our study, knockdown of the TLR9 pathway in MSCs reduces the effectiveness of the cells in enhancing granulopoiesis, suggesting a role for DNA sensing in regulating demand adaptive lineage switching. Increased levels of GC-rich DNA can occur either as a consequence of dying eukaryotic cells releasing mitochondrial DNA,66 or from bacteria. P aeruginosa, a common opportunistic pathogen, has an overabundance (67% total genomic DNA) of GC sequences67 and has a marked ability to stimulate TLR9.68 TLR9, in turn, has been shown to enhance the ability of mesenchymal stromal cells to migrate toward sites of damage and inflammation.69,70 In this study, we demonstrated that both MSCs and CpG-MSCs have the ability to migrate to the bone marrow after radiation injury; however, we did not observe significant migratory superiority when MSCs were preconditioned with CpG. Given this, we determined that the observed advantage of CpG-MSCs on granulopoiesis was not driven by enhanced migration to the bone marrow, but rather, that preconditioning bone marrow MSCs with CpG can alter the MSC secretome to promote granulopoiesis when challenged with a pathogen. Multiplex analysis of conditioned media from MSCs and CpG-MSCs additionally identified an increase in the amount of PDGF-AA in the conditioned media of CpG-MSCs compared with MSCs. Intriguingly, PDGF-BB, which binds to both α and β isoforms of the PDGF receptor, has been shown to be able to stimulate CFU-GM proliferation in vitro with an effect size that was comparable to stimulation with IL-3, IL-6, GM-CSF, and acidic fibroblast growth factor.49 The results of our study suggest that PDFG-AA, which binds only to the α isoform of the PDGF receptor, may also play a role in stimulating granulopoiesis in vivo. Although our model was xenogeneic in nature, and some soluble factors such as GM-CSF and IL-3 are not species cross reactive,71 human and mouse G-CSF have complete biological cross reactivity,42 and human IL-6 has been demonstrated to be active on murine cells.72,73 Notably, recombinant PDGF-AA has also been used in murine models with demonstration of species cross reactivity.74 Therefore, further characterization of the role of PDGF-AA, and the relative contributions of other contents of the conditioned media, warrants further investigation.
Despite increased neutrophils, however, we did not observe a reduction in organ bacterial count. Instead, mice that received CpG-MSCs had less organ damage. This is consistent with prior studies demonstrating that MSCs can have anti-inflammatory75 and pro-resolving76,77 effects on unrestrained immune activation, and therapeutically addresses the neutrophil dysregulation that occurs in severe sepsis.78 NETs have been shown to be beneficial in pathogen containment,79 yet also detrimental by causing tissue injury and inflammation.45 In sepsis-associated acute respiratory distress syndrome, increased plasma NETs were associated with mortality and the severity of lung injury.80 Decreasing but not eliminating NET formation with DNase I has additionally been shown to reduce lung injury and improve survival. Taken together, our study demonstrates that a fine balance between augmenting granulopoiesis but reducing tissue injury can be struck with the use of MSCs.
Currently, the American Society of Clinical Oncology has a limited arsenal of white blood cell growth factors that are recommended for use in the setting of neutropenia and its associated complications.81 Beyond the population of patients receiving iatrogenic radiation injury, the Department of Health and Human Services and the Department of Defense have appropriated funds to create a national stockpile of therapeutics to respond to a radiation public health disaster, such as Chernobyl or Fukushima.82,83 Agents included in the stockpile currently consist of chelating agents, G-CSF, and GM-CSF84 ; however, there remains a dire need to create additional medical countermeasures for radiation toxicity. Here we present 1 such therapeutic candidate with a favorable clinical safety profile85,86 for the treatment of radiation-associated neutropenia to reduce mortality from complicating sepsis, with dual effects of augmenting granulopoiesis while reducing organ damage.
For original data, please contact jng9@bwh.harvard.edu.
Acknowledgments
The authors thank Bruce Levy, Rebecca Baron, and Issac Chiu for their scientific input, and Gerald Pier for his scientific input and for providing the Pseudomonas-GFP used in these experiments.
Imaging, consultation, and/or services were performed in the Neurobiology Imaging Facility at Harvard Medical School. This facility is supported, in part, by the HMS/BCH Center for Neuroscience Research as part of a National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke P30 Core Center grant (NS072030). This study was supported by grants from the NIH/National Heart, Lung, and Blood Institute (T32HL007633-29), NIH/National Institute of Allergy and Infectious Diseases (U01AI138318), and NIH/National Institute of General Medical Sciences (R01GM118456 and R01GM136804).
Authorship
Contribution: J.N. participated in the experimental design, carried out most of the experiments, and drafted the manuscript; F.G. and L.A.C. carried out the survival and bronchoalveolar lavage experiments and helped with all the in vivo experiments; A.E.M. participated in the colony-forming assays; S.G. performed the MTT assay and prepared the MSCs for animal injection; M.-Y.K. helped with silencing of TLR9 and development of the phagocytosis assays; K.T.W. independently scored bone histology sections; J.K. and X.L. helped with the in vivo experiments; B.K. helped with the NETosis assays; A.M. contributed significant scientific input; M.A.P. and J.A.L. conceived to the study, participated in its design and coordination, and helped to draft the manuscript; and all authors have read and approved the final manuscript.
Conflict-of-interest disclosure: A.M. has received honoraria from Blueprint Medicines, Roche, and Incyte, and receives research support from Janssen and Actuate Therapeutics. The remaining authors declare no competing financial interest.
Correspondence: James A. Lederer, Division of Surgery, Brigham and Women’s Hospital, 75 Francis St, Boston, MA 02215; e-mail: jlederer@bwh.harvard.edu; or Mark A. Perrella, Division of Pulmonary and Critical Care Medicine, Brigham and Women's Hospital, 75 Francis St, Boston, MA 02115; e-mail: mperrella@rics.bwh.harvard.edu.

