

Key Points
In selected mouse strains, platelet alloimmunization regularly transitions to autoantibody production and thrombocytopenia.
PTP is an autoimmune disorder and can be mimicked in mice.

Abstract
Posttransfusion purpura (PTP) is an uncommon but life-threatening condition characterized by profound thrombocytopenia occurring ∼1 week after a blood transfusion. The hallmark of PTP is a potent immunoglobulin G antibody specific for a transfused platelet-specific alloantigen, usually located on glycoprotein IIb/IIIa (GPIIb/IIIa; αIIb/β3 integrin). It is widely thought that this alloantibody somehow causes the thrombocytopenia, despite absence from host platelets of the alloantigen for which it is specific. In studies described here, we found that cross-strain platelet immunization in mice commonly induces GPIIb/IIIa-specific alloantibodies combined with platelet-specific autoantibodies and varying degrees of thrombocytopenia, and we identified 1 strain combination (129S1Svlm/PWKPhJ) in which 95% of immunized mice made both types of antibody and developed severe thrombocytopenia. There was a strong inverse correlation between autoantibody strength and platelet decline (P < .0001) and plasma from mice that produced autoantibodies caused thrombocytopenia when transfused to syngeneic animals, arguing that autoantibodies were the cause of thrombocytopenia. The findings define a model in which a routine alloimmune response to platelets regularly transitions to an autoimmune reaction capable of causing severe thrombocytopenia and support the hypothesis that PTP is an autoimmune disorder.
Introduction
It has long been known that immunization against red blood cell (RBC) alloantigens following transfusion is sometimes accompanied by production of RBC-specific autoantibodies.1-3 Usually, affected patients are asymptomatic but severe4 and even fatal5 hemolytic episodes have been recorded. Single case reports suggest that a similar phenomenon occurs in some patients mounting an immune response against transfused human platelet alloantigens (HPAs)6-10 and it has been proposed that, owing to the small mass of circulating platelets, these “companion” autoantibodies can cause thrombocytopenia as part of an uncommon, but life-threatening, complication of blood transfusion designated “posttransfusion purpura” (PTP).10 In both of these circumstances, clinical and serologic findings are consistent with the possibility that a normal immune response against a transfused alloantigen somehow transitions to an autoimmune one capable of destroying autologous cells. Mouse models can be useful for characterizing the alloresponse against transfused RBCs11,12 and platelets.13-16 In this report, we identify conditions under which cross-strain immunization of mice with platelets consistently leads to production of alloantibodies that recognize glycoprotein IIb/IIIa (GPIIb/IIIa) from the immunizing strain as well as autoantibodies capable of causing severe thrombocytopenia. The model appears to recapitulate findings seen in human patients with PTP and should facilitate further studies to define the molecular basis for a transition from alloimmunity to autoimmunity in this condition.
Methods
Mice
C57Bl/6J (C57) 129S1/SvlmJ (129), SPRET/EiJ (SPRET), and PWK/PhJ (PWK) mouse strains were obtained from The Jackson Laboratory (Bar Harbor, ME) and were bred under pathogen-free conditions. C57 and 129 are widely used, well-characterized strains that possess the H-2b major histocompatibility complex (MHC) haplotype. The SPRET and PWK strains are derived from mice wild-caught in different parts of Europe and their MHC haplotypes are undefined.
Immunizations
Mouse platelets were isolated by centrifuging citrated whole blood through a Histopaque (Millipore Sigma, St. Louis, MO) gradient (density = 1.077). Washed platelets were suspended in a 1:1 ratio of Sigma Adjuvant System (Millipore Sigma) and 0.2 mL (1 × 108 platelets) was injected intraperitoneally weekly for 5 weeks. EDTA blood samples (Microvette; Sarstedt, Numbrecht Germany) were obtained from the submandibular vein prior to immunization and 2 days after each immunization. Complete blood counts were performed using the Animal Blood Counter (Scil, Gurnee, IL). An end-of-study blood sample, drawn from the vena cava, was collected in sodium citrate.
Serologic studies
For platelet-associated immunoglobulin G (IgG; PAIgG) measurements, washed platelets (1 × 106) were combined with 1/100 diluted fluorescein isothiocyanate (FITC)–labeled goat anti-mouse IgG (Fc-specific) F(ab′)2 (Jackson Immunoresearch, West Grove PA) in a 100-μL total volume. After incubation for 45 minutes, samples were diluted and bound secondary antibody was detected using an Accuri C6 flow cytometer (Becton Dickenson, San Jose, CA). For measurement of alloantibody and autoantibody, platelets from mice of the donor (for alloantibodies) and recipient (for autoantibody), strains (5 × 106) were combined with 10 μL of test plasma in a final volume of 50 μL. After incubation for 1 hour at room temperature, platelets were washed and suspended in 50 μL of 1/100 diluted anti-mouse IgG Fc F(ab′)2 and bound secondary antibody was measured as previously described. Antibody strength was expressed as the ratio of the median fluorescent intensity (MFI) signal obtained with a postimmunization plasma sample to the signal obtained with a preimmunization sample studied simultaneously.
Detection of MHC antibodies
Splenic T cells were marked for detection of MHC-specific antibodies as previously described17 with slight modifications. Total splenocytes (1 × 105) from the donor strain mice were combined with preimmune and final bleed samples (10 μL) in a 50-μL volume. Following incubation for 1 hour, the cells were washed and treated with FITC-labeled rat anti-mouse CD3 (Thermo Fisher Scientific, Waltham, MA), phycoerythrin-labeled rat anti-mouse CD45R (B220; ThermoFisher), and allophycocyanin-labeled goat anti-mouse IgG Fc F(ab′)2, and incubated for an additional 45 minutes. Samples were diluted and analyzed by flow cytometry. IgG binding was measured on cells gated as CD3+ (FITC) and CD45R− (phycoerythrin).
Identification of targets recognized by alloantibody
Immunoprecipitation studies were as previously described18 with slight modifications. Briefly, platelet surface glycoproteins were labeled with biotin by combining equal volumes of washed platelets (4 × 109/L) and 10 mM EZ Link NHS-LC biotin (Thermo Fisher Scientific) for 30 minutes in phosphate-buffered saline. Washed platelets were sensitized with terminal-bleed plasma (60 μL) or MWReg30 (anti-mouse CD41) (BD Biosciences, San Jose, CA), then washed and lysed. Sensitizing antibodies along with bound surface proteins were captured on protein G sepharose beads and washed. Captured proteins were eluted by boiling in sodium dodecyl sulfate (SDS) loading buffer, subjected to SDS–polyacrylamide gel electrophoresis, transferred to polyvinylidene fluoride, and western blotted with horseradish peroxidase–streptavidin.
Statistics
Spearman correlation analysis was used to assess the association of the strength of autoantibody and platelet nadirs, and was calculated using GraphPad Prism (v6.05). The Student t test was used for comparison of 2 groups.
Study approval
Animal studies were approved by the institutional animal care and use committee of the Medical College of Wisconsin (Milwaukee, WI).
Results
Selection of mouse strains for platelet immunizations
In humans, GPIIb/IIIa (αIIb/β3 integrin) and GPIbα are the most common targets for platelet-specific alloantibodies19,20 and autoantibodies.21 More than 90% of alloantibodies associated with the human disorder, PTP, recognize these 2 glycoprotein complexes.22 To identify mouse strains that might be capable of producing alloantibodies that recognize these targets, we used the Mouse Genome database of the Wellcome Sanger Institute (https://www.sanger.ac.uk/sanger/Mouse_SnpViewer/rel-1303) to identify the amino acid composition of these proteins in various mouse strains and, on the basis of findings made, selected the strains C57Bl/6J (C57), 129S1/SvlmJ (129), SPRET/EiJ (SPRET), and PWK/PhJ (PWK) for these studies. Table 1 shows amino acid residues at which the 4 strains differ from one another in the extracellular domains of GPIIb, GPIIIa, and GPIbα to which potential alloantibodies would be expected to bind. The widely used strains C57 and 129 differ by only 2 aa in extracellular GPIIb but PWK and SPRET differ by a total of 19 residues distributed among the 3 glycoproteins and from the reference strain C57 by 9- and 18-aa residues, respectively. Increased disparities in amino acid composition of these glycoproteins in PWK and SPRET relative to each other and to the C57 and 129 strains are likely a consequence of the relatively recent, wild-derived origin of PWK and SPRET mice.23
Amino acid residues at which extracellular domains of GPIIb, GPIIIa, and GPIbα are predicted to differ among 4 mouse strains
| GPIIb* | GPIIIa* | GP1bα* | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mouse strain | 37 | 111 | 179 | 392 | 805 | 933 | 976 | 103 | 297 | 677 | 323 | 374 | 381 | 382 | 422 | 429 | 432 | 444 | 459 | 490 | 493 | 496 | 545 | ||
| C57 | E | G | A | P | R | V | R | A | H | A | A | K | P | S | T | H | I | K | L | T | H | T | T | ||
| 129 | — | — | — | — | S | A | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | ||
| PWK | V | — | — | — | S | A | Q | S | — | — | — | — | T | — | — | — | — | — | — | N | P | — | I | ||
| SPRET | — | S | D | S | S | A | — | — | R | S | T | E | T | T | A | P | T | E | P | N | — | I | — | ||
| GPIIb* | GPIIIa* | GP1bα* | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mouse strain | 37 | 111 | 179 | 392 | 805 | 933 | 976 | 103 | 297 | 677 | 323 | 374 | 381 | 382 | 422 | 429 | 432 | 444 | 459 | 490 | 493 | 496 | 545 | ||
| C57 | E | G | A | P | R | V | R | A | H | A | A | K | P | S | T | H | I | K | L | T | H | T | T | ||
| 129 | — | — | — | — | S | A | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | ||
| PWK | V | — | — | — | S | A | Q | S | — | — | — | — | T | — | — | — | — | — | — | N | P | — | I | ||
| SPRET | — | S | D | S | S | A | — | — | R | S | T | E | T | T | A | P | T | E | P | N | — | I | — | ||
— indicates identity with C57.
Numbers denote amino acid positions at which extracellular domains of GPIIb, GPIIIa, and GPIbα of the 4 mouse strains differ from one another. C57 was chosen as the reference strain. Letters assigned to the other 3 strains indicate amino acids that differ from C57. Amino acid 1 starts with the ATG of the translation start codon for each of the expressed proteins. Amino acids are designated by the conventional letter code. No intracellular polymorphisms have been reported in these glycoproteins.
Representative hematologic and serologic responses to platelet immunization
A total of 57 intraperitoneal immunizations (48 cross-strain and 9 syngeneic) involving different mouse combinations designated “groups 1-8” (Table 2) were performed with platelets from 3 of the 4 strains to determine which combinations induced platelet-specific immune responses with or without associated thrombocytopenia. SPRET platelets were not used for immunization because of problems encountered in breeding a sufficient number of mice for platelet harvesting. We will refer to antibodies that recognize only platelets of the immunizing strain as “alloantibodies” and those that recognize platelets from both donor and recipient strains as “autoantibodies.” Figure 1 illustrates types of responses observed. In Figure 1A (PWK donor, C57 recipient), an alloantibody reactive with immunizing PWK platelets appeared at week 3. However, plasma autoantibody was not identified, PAIgG (2.20 ratio) was slightly elevated, and platelet levels were stable. In Figure 1B (129 donor, PWK recipient), a strong alloantibody was first detected at week 3. At week 4, platelets dropped by ∼76% and autoantibody and PAIgG (2.55 ratio) became elevated. In Figure 1C (PWK donor, 129 recipient), alloantibody appeared at week 2, and autoantibody at week 3 in association with a 96% drop in the platelet count and an increase in PAIgG (3.70 ratio). Figure 1D shows that after injection of syngeneic platelets, neither alloantibodies nor autoantibodies were detected, and there was no decline in platelet level.
Donor-recipient combinations used for platelet immunizations
| Group | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Donor | C57 | 129 | PWK | C57 | 129 | C57 | C57 | 129 | 129 | PWK | PWK |
| Recipient | C57 | 129 | PWK | 129 | C57 | SPRET | PWK | SPRET | PWK | C57 | 129 |
| n | 4 | 3 | 2 | 4 | 3 | 6 | 4 | 6 | 6 | 6 | 13 |
| Group | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Donor | C57 | 129 | PWK | C57 | 129 | C57 | C57 | 129 | 129 | PWK | PWK |
| Recipient | C57 | 129 | PWK | 129 | C57 | SPRET | PWK | SPRET | PWK | C57 | 129 |
| n | 4 | 3 | 2 | 4 | 3 | 6 | 4 | 6 | 6 | 6 | 13 |
Four strains of mice were immunized (recipients) with platelets from 3 strains (donors) in 8 groups.
n, number of mouse pairs in each combination.

Representative patterns of response following platelet immunization. Platelet counts (109/L; ○) are shown on the left ordinate and alloantibody (AlloAb; □) and autoantibody (AutoAb; △) responses on the right. Alloantibody responses are expressed as the ratio of the median fluorescence signal obtained postimmunization to the signal obtained with pretreatment plasma using platelets of the immunizing strain as targets in flow cytometry. Autoantibodies are similarly expressed using platelets from the recipient strain as antibody targets. Weeks of immunization are shown on the abscissa. (A) A C57 mouse immunized with PWK platelets produced a strong alloantibody but no detectable autoantibody, and platelets were unaffected. (B) A PWK mouse immunized with 129 platelets produced a strong alloantibody and a moderately strong autoantibody; platelets declined ∼76% from the maximum level. (C) A 129 mouse immunized with PWK platelets produced strong alloantibodies and autoantibodies and experienced a platelet decline of 96%. (D) A 129 mouse immunized with 129 platelets produced neither alloantibodies nor autoantibodies and experienced a platelet decline of 23% from the maximum value.
Representative patterns of response following platelet immunization. Platelet counts (109/L; ○) are shown on the left ordinate and alloantibody (AlloAb; □) and autoantibody (AutoAb; △) responses on the right. Alloantibody responses are expressed as the ratio of the median fluorescence signal obtained postimmunization to the signal obtained with pretreatment plasma using platelets of the immunizing strain as targets in flow cytometry. Autoantibodies are similarly expressed using platelets from the recipient strain as antibody targets. Weeks of immunization are shown on the abscissa. (A) A C57 mouse immunized with PWK platelets produced a strong alloantibody but no detectable autoantibody, and platelets were unaffected. (B) A PWK mouse immunized with 129 platelets produced a strong alloantibody and a moderately strong autoantibody; platelets declined ∼76% from the maximum level. (C) A 129 mouse immunized with PWK platelets produced strong alloantibodies and autoantibodies and experienced a platelet decline of 96%. (D) A 129 mouse immunized with 129 platelets produced neither alloantibodies nor autoantibodies and experienced a platelet decline of 23% from the maximum value.
Alloantibody responses
As shown in Figure 2A, alloantibodies were detected in each of 48 interstrain (groups 2-8) but not in any of 9 syngeneic (group 1) immunizations. To assure comparability between measurements made in different animals, the strength of a response was expressed as the ratio of the MFI signal obtained following immunization to that obtained with a preimmunization sample tested simultaneously. Alloresponses were weak in immunizations between the MHC-identical (H-2b) strains C57 and 129 (group 2; average MFI ratio, 9.1) in which GPIIb/IIIa and GPIb differ in their extracellular domains only by a total of 2-aa residues in GPIIb but were on average about 13 times stronger in combinations involving at least 1 wild-derived mouse strain (PWK or SPRET) where donor and recipient mice differed by 7 to 18 aa (groups 3-8).
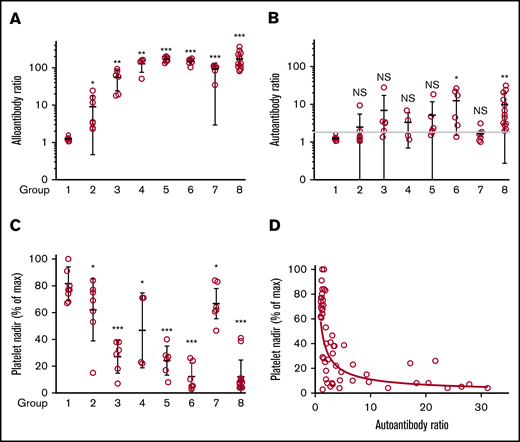
Alloantibody, autoantibody, and platelet responses observed in all immunizations. Identity of strains used as platelet donors and recipients and number of mice in each of the 8 groups are shown in Table 2. (A) Alloantibody responses were strongest in immunizations involving at least 1 wild-derived mouse strain (PWK, SPRET, groups 3-8). Much weaker alloresponses were seen in reciprocal immunizations involving MHC-matched C57 and 129 (group 2) that differed by only 2 aa in GPIIb. No significant responses were seen following immunization with strain-identical platelets (group 1). *P < .05; **P < .01; ***P < .001. (B) Autoantibody responses were identified in 33 of 48 mice injected with platelets from other strains (groups 2-8). Using the mean + 3 standard deviation (1.8) obtained with the strain-identical immunizations as the cutoff for a "positive" reaction. (C) Platelet nadirs (percentage of maximum platelet count) in mice of groups 1 through 8. (D) There was an inverse correlation between platelet autoantibody levels and platelet nadir (Spearman coefficient P < .0001; r = −0.75). NS, not significant.
Alloantibody, autoantibody, and platelet responses observed in all immunizations. Identity of strains used as platelet donors and recipients and number of mice in each of the 8 groups are shown in Table 2. (A) Alloantibody responses were strongest in immunizations involving at least 1 wild-derived mouse strain (PWK, SPRET, groups 3-8). Much weaker alloresponses were seen in reciprocal immunizations involving MHC-matched C57 and 129 (group 2) that differed by only 2 aa in GPIIb. No significant responses were seen following immunization with strain-identical platelets (group 1). *P < .05; **P < .01; ***P < .001. (B) Autoantibody responses were identified in 33 of 48 mice injected with platelets from other strains (groups 2-8). Using the mean + 3 standard deviation (1.8) obtained with the strain-identical immunizations as the cutoff for a "positive" reaction. (C) Platelet nadirs (percentage of maximum platelet count) in mice of groups 1 through 8. (D) There was an inverse correlation between platelet autoantibody levels and platelet nadir (Spearman coefficient P < .0001; r = −0.75). NS, not significant.
Autoantibody responses
Platelet-reactive autoantibodies were evaluated in 2 ways. In one, reactions of plasma against platelets from animals of the same strain were measured; in the other, IgG bound to platelets of immunized animals (PAIgG) was determined. There was a general correlation between results obtained by the 2 methods. However, because of difficulties in obtaining sufficient platelets for PAIgG measurements in animals with severe thrombocytopenia, the ratio of the MFI signal obtained with a postimmunization sample to that obtained with a stored preimmunization sample was used to define autoantibody activity. Results are summarized in Figure 2B. In control animals injected with strain-identical platelets (group 1), the ratio of the MFI signal obtained with postimmunization to preimmunization plasma ranged from 1.0 to 1.6 (mean, 1.24; standard deviation, 0.17). Using the mean + 3 standard deviation (1.8) obtained with the strain-identical immunizations as the cutoff for a “positive” reaction, 33 of 48 mice (69%) immunized with platelets from a different strain produced autoantibodies that recognized syngeneic platelets (Figure 2B). When defined as the ratio of the fluorescent signals obtained with postimmunization and preimmunization samples, the strength of autoantibodies was much weaker than that of alloantibodies, possibly because of the former being removed from the circulation as they react with and cause destruction of autologous platelets.
Platelet levels
After 2 immunizations and prior to development of alloantibodies or autoantibodies, platelet levels increased by 15% to 100%. Comparable increases, persisting for up to 5 injections, were seen in animals given adjuvant alone, indicating that this rise is a nonspecific response to adjuvant-induced inflammation.24,25 RBC counts declined ∼10%, attributable to blood draws, and white blood cell levels increased by an average of 16%. Platelet decreases in immunized animals were expressed as a percentage of the maximum platelet count obtained postimmunization. In 9 mice injected with strain-identical platelets (group 1), platelets declined by an average of 18% (range, 0% to 33%) and in 7 mice injected with MHC-identical, strain-disparate platelets (group 2), the average decline was 38% (range, 16% to 85%) (Figure 2C). However, in 41 mice injected with platelets from animals that were both strain and MHC disparate (groups 3-8), platelets declined by an average of 73% (range, 16% to 97%; median, 77%). The most consistent and severe declines occurred in PWK mice immunized with 129 platelets and vice versa (groups 6 and 8; N = 19) in which the mean platelet count decline was 88% (range, 59% to 97%; median, 92%). At the other extreme, in C57 mice immunized with PWK platelets (group 7; N = 6), the average platelet count drop was only 33% (range, 16% to 53%; median, 37%), despite production of moderately strong alloantibodies by these mice.
Decreases in platelet levels correlated inversely with autoantibody activity
Figure 2D, containing data from 48 animals immunized with strain-disparate platelets, illustrates a highly significant, inverse relationship between platelet levels and the strength of autoantibodies. As shown in supplemental Table 1, mice with platelet nadirs that fell into the highest, middle, and lowest third of all values had autoantibody values that averaged 1.59, 5.60, and 12.45, respectively: differences that were highly significant. To demonstrate that autoantibodies detected were capable of destroying autologous platelets, end-of-study citrated plasma samples (100 μL) from 5 mice with detectable autoantibodies associated with thrombocytopenia and 3 mice with no detectable autoantibodies or thrombocytopenia were transfused to syngeneic mice. As shown in Figure 3, platelet counts obtained 1 day after infusion were significantly lower in mice given autoantibody-containing plasma.
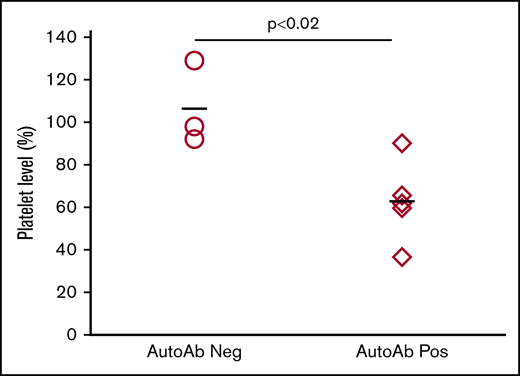
Autoantibodies cause platelet clearance. Individual “end-of-study” plasma samples from selected mice were injected IV into individual syngeneic mice. Platelet counts measured 24 hours after injection were compared to preinjection counts and are expressed as a percentage of the preinjection count. Platelet levels in mice injected with plasma samples with no detectable autoantibodies (○) remained unaffected. Platelet levels in mice injected with plasma samples containing autoantibodies and associated with thrombocytopenia (♢) decreased significantly (P < .02). The bars depict the average of all animals in the group.
Autoantibodies cause platelet clearance. Individual “end-of-study” plasma samples from selected mice were injected IV into individual syngeneic mice. Platelet counts measured 24 hours after injection were compared to preinjection counts and are expressed as a percentage of the preinjection count. Platelet levels in mice injected with plasma samples with no detectable autoantibodies (○) remained unaffected. Platelet levels in mice injected with plasma samples containing autoantibodies and associated with thrombocytopenia (♢) decreased significantly (P < .02). The bars depict the average of all animals in the group.
Targets recognized by alloantibodies and autoantibodies on platelets
Specificity of 15 arbitrarily chosen alloantibodies was studied by observing their ability to capture membrane proteins from biotinylated platelets of the mouse strain used for immunization. Twelve of these precipitated GPIIb/IIIa from donor strain platelets used for immunization as illustrated for a group 3 (C57 to SPRET) and a group 4 (C57 to PWK) immunization in Figure 4. The latter sample was unique in also precipitating a band of molecular mass expected for GPIb (170 kDa). A band correlating with the expected molecular weight of class I MHC (45 kDa) was not found in any of the 15 studies. Attempts to precipitate proteins targeted by autoantibodies on syngeneic platelets were unsuccessful, possibly owing to the weakness of these antibodies.
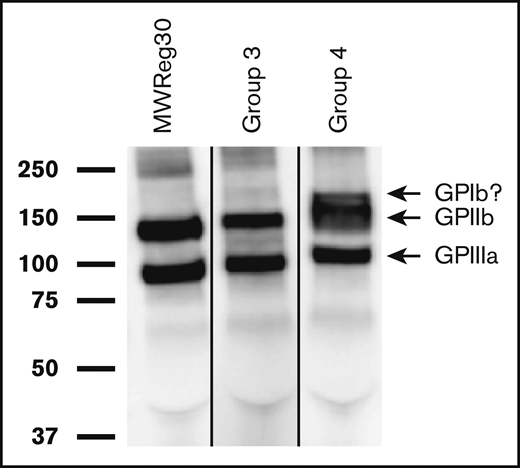
Alloantibodies recognize GPIIb/IIIa on platelets from the strain used for immunization. Biotin-labeled platelet membrane proteins from the mouse strain used for immunization were immunoprecipitated with alloantibodies from immunized mice, subjected to SDS–polyacrylamide gel electrophoresis and detected by western blotting with horseradish peroxidase–streptavidin as described in “Methods.” Molecular weight standards are indicated on the left. Shown here are representative results obtained in 2 of 12 studies involving immunization of SPRET (group 3) and PWK (group 4) mice with C57 platelets. Bands identified correlate with those precipitated by monoclonal rat anti-mouse antibody MWReg30 specific for mouse GPIIb/IIIa (left gel). The extra band above the GPIIb band obtained with alloantibody from group 4 has the apparent molecular weight expected for GPIb (170 kDa) but was not further investigated. The lanes were run on the same gel but were noncontiguous.
Alloantibodies recognize GPIIb/IIIa on platelets from the strain used for immunization. Biotin-labeled platelet membrane proteins from the mouse strain used for immunization were immunoprecipitated with alloantibodies from immunized mice, subjected to SDS–polyacrylamide gel electrophoresis and detected by western blotting with horseradish peroxidase–streptavidin as described in “Methods.” Molecular weight standards are indicated on the left. Shown here are representative results obtained in 2 of 12 studies involving immunization of SPRET (group 3) and PWK (group 4) mice with C57 platelets. Bands identified correlate with those precipitated by monoclonal rat anti-mouse antibody MWReg30 specific for mouse GPIIb/IIIa (left gel). The extra band above the GPIIb band obtained with alloantibody from group 4 has the apparent molecular weight expected for GPIb (170 kDa) but was not further investigated. The lanes were run on the same gel but were noncontiguous.
Reactions of alloantibodies with T cells
Despite failure to demonstrate reactions of alloantibodies with class I MHC molecules on platelets, plasma samples from 27 of 28 mice immunized with MHC-incompatible platelets (groups 3-8) contained IgG antibodies that recognized splenic T cells from the strain used for immunization (average postmmunization to preimmunization MFI ratio, 29.3). Platelets used for immunization were leukocyte-poor (<100/μL) but not leukocyte-free, so MHC antigens on platelets, leukocytes, or both could have induced T-cell–reactive antibodies, possibly specific for class I MHC or other T-cell membrane proteins that differ in amino acid composition between strains. For the purpose of this study, we addressed only the question of whether MHC-specific antibodies significantly affected alloantibody reactions against immunizing platelets (Figure 2A) by comparing the strength of class I MHC expression on platelets and CD3+ spleen cells of 129 strain mice. As shown in supplemental Figure 1, H2kb was strongly expressed on CD3+ spleen cells as expected but was barely detectable on 129 strain platelets, suggesting that reactions of MHC-specific alloantibodies provided only a minor contribution to alloantibody reactions shown in Figure 2A. Several earlier reports have also suggested that class I MHC antigens are weakly expressed on platelets of some mouse strains.16,26
Discussion
Studies of the pathogenesis of autoimmune hemolytic anemia and thrombocytopenia going back almost 50 years have used animal models to show that cross-species and cross-strain immunization with blood cells sometimes induces production of antibodies specific not only for the immunizing “foreign” cells but also for corresponding autologous cells as well. Early reports showed that intraperitoneal injection of mice with rat RBCs regularly led to production of antibodies that recognized both rat and mouse RBCs27-30 and that immunization of mice with rat platelets induced platelet-specific autoantibodies and moderate thrombocytopenia.31,32 We are aware of only 2 previous studies in which autoimmune thrombocytopenia appears to have developed following platelet immunization between members of the same species. In 1965, Baldini described occasional, severe, and prolonged thrombocytopenia in dogs following repeated platelet transfusions from other randomly selected animals and attributed this to induction of autoantibodies.33 In the late 1970s, Gengozian and coworkers found that weekly intramuscular injection of platelets from 1 strain of marmosets to another in complete Freund adjuvant led to production of platelet-reactive alloantibodies and autoantibodies, profound thrombocytopenia, and a high rate of mortality due to bleeding.34,35 Each study involving cross-species or cross-strain platelet immunization in animals made note of the similarity of findings made to the human disorder, PTP.31,33,34
Our studies are the first to demonstrate induction of alloantibodies, autoantibodies, and thrombocytopenia following cross-strain platelet immunization in mice. All animals immunized with platelets from a mouse of a different strain produced IgG alloantibodies that recognized donor platelets, and approximately two-thirds also made autoantibodies capable of lowering platelet counts. Immunization by the intraperitoneal route was necessary to induce the full range of hematologic and serologic findings because we found that platelets injected between strains IV induce strong platelet-specific alloantibodies but no detectable autoantibodies and have no effect on platelet levels (supplemental Figure 2). Similar findings have been made in other studies involving interstrain platelet transfusions.13-16 The dominant target for alloantibodies produced by our mice was GPIIb/IIIa (αIIb/β3 integrin) but plasma from 1 of 15 mice also recognized a protein with molecular mass consistent with GPIb. Within the limits of this study (3 immunizing strains, 4 recipient strains), animals that differed from the immunizing strain by 7 to 18 aa in the extracellular domains of GPIIb/IIIa and GPIb (groups 3-8) produced much stronger alloantibodies than those with only 2-aa differences (group 2). It was found empirically that the most consistent production of strong alloantibodies together with autoantibodies capable of causing severe thrombocytopenia occurred when 129 mice were immunized with platelets from wild-derived PWK mice (group 8) and vice versa (group 6). These 2 strains differ by a total of 7 aa in extracellular domains of GPIIb/IIIa and GPIbα (Table 1).
PTP, first defined as a clinical entity in 1961,36 is characterized by severe, often life-threatening, thrombocytopenia occurring ∼1 week after blood transfusion. Serologic studies invariably demonstrate high-titer IgG antibodies specific for 1 or more HPAs, usually HPA-1a, found in ∼98% of healthy individuals. Platelet levels eventually return to normal in most cases, but profound thrombocytopenia sometimes persists for 3 to 4 weeks and mortality is ∼15%.10,37,38 It has generally been assumed that the potent HPA-specific alloantibody typically found in PTP somehow causes platelet destruction even though platelets from affected individuals lack the antigen for which it is specific and antibody usually remains detectable after recovery. To explain these seeming contradictions, it was suggested that a soluble form of alloantigen transfused with blood might react with antibody to form immune complexes capable of causing thrombocytopenia36 or might bind directly to autologous platelets, causing them to become an antibody target,39 but both of these proposals would require the postulated soluble antigen to persist in the circulation for many weeks in the face of a potent alloantibody that would be expected to promote its clearance. An alternative explanation, that autologous platelets are destroyed by autoantibody produced concomitantly with the more prominent alloantibody, was suggested by studies performed in single patients with PTP in whom apparent platelet-reactive autoantibodies were demonstrated during the acute phase of the disease.6-10 In several particularly convincing studies, autoantibody activity was confirmed by showing that patient platelets obtained postrecovery reacted with serum collected during the thrombocytopenic phase.7,8,10 Thus, emerging clinical evidence is consistent with the possibility that PTP is an autoimmune disorder caused by self-reactive antibodies produced as a by-product of the immune response against a platelet alloantigen.
In a recent report, we hypothesized how this transition from alloimmunity to autoimmunity might take place.10 Typically, a blood cell alloantigen is created by a single amino acid polymorphism in a membrane protein that causes it to be recognized as “foreign” following transfusion to a host that lacks the antigen (Figure 5). An alloantibody specific for such an antigen is critically dependent on the polymorphic amino acid for binding, but adjacent invariant residues also make important contributions to the binding energy.40-43 Thus, every alloantibody recognizes invariant amino acid residues in the vicinity of the polymorphic one. The immune response leading to production of an IgG antibody involves somatic hypermutation in which amino acids that make up the antigen-binding site (complementarity determining region [CDR]) are randomly mutated and B cells with receptor modifications that increase affinity for the target are selected for proliferation.44 It is not difficult to envision how, in the course of an alloresponse, mutation of a single amino acid or a very small number of amino acids in the alloantibody CDR could modify its specificity in such a way that it recognizes nonpolymorphic amino acids with higher affinity and becomes less dependent or independent of the single polymorphic one for binding. Immunoglobulins produced by such a B-cell clone would recognize invariant amino acid residues found on both the immunizing antigen and its autologous counterpart and would resemble autoantibodies described in individual patients with PTP7,10 and in patients mounting an immune response against an RBC alloantigen.1-5 The mechanism we propose for induction of autoimmunity in PTP can be considered to be a special example of “epitope spreading,” a well-recognized immunologic phenomenon in which the immune response to an exogenous antigen is redirected as the result of antigen “cross-reactivity” or 1 of several other mechanisms,45-47 including chance CDR restructuring that occurs in the course of somatic hypermutation.48
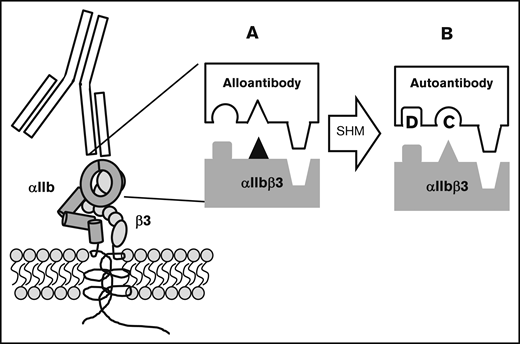
Proposed mechanism for conversion of an alloantibody to an autoantibody during somatic hypermutation in an evolving humoral immune response. Blocks depict the antibody-antigen interface. (A; ▲) The polymorphic amino acid that determines an alloantigen and initially induces an alloantibody that recognizes this and adjacent nonpolymorphic residues. (B) By chance, somatic hypermutation (SHM) creates additional mutations (C and D) in the antibody CDR. Mutation C abolishes affinity for the amino acid that determines the alloantigen and mutation D enhances affinity for an adjacent nonpolymorphic amino acid. The resulting autoantibody binds to the same region on the counterpart autologous protein with sufficient affinity to lead to platelet destruction.
Proposed mechanism for conversion of an alloantibody to an autoantibody during somatic hypermutation in an evolving humoral immune response. Blocks depict the antibody-antigen interface. (A; ▲) The polymorphic amino acid that determines an alloantigen and initially induces an alloantibody that recognizes this and adjacent nonpolymorphic residues. (B) By chance, somatic hypermutation (SHM) creates additional mutations (C and D) in the antibody CDR. Mutation C abolishes affinity for the amino acid that determines the alloantigen and mutation D enhances affinity for an adjacent nonpolymorphic amino acid. The resulting autoantibody binds to the same region on the counterpart autologous protein with sufficient affinity to lead to platelet destruction.
The mouse model described here mimics the human condition PTP in many respects, including kinetics of the immune responses to platelet immunization, thrombocytopenia correlated with an alloimmune response against a platelet-specific antigen usually located on GPIIb/IIIa, and the possibility that certain MHC mismatches (eg, PWK/129) favor the alloresponse and autoresponses to a transfused antigen, as is true of the immune response to the alloantigen HPA-1a in humans.49-51 A contrast to the human disorder is that, in the mouse model, platelets providing the immunogenic stimulus were injected intraperitoneally with adjuvant, rather than being given IV. PTP is not a common disorder and it is likely that a very large number of transfusions might be needed to induce it in a mouse the “natural” way. Extravascular injection of platelets with adjuvant may be necessary to overcome normal tolerance mechanisms and induce an immune response sufficiently robust for autoantibody induction to occur in a manageable number of animals.
Tools now available for characterizing the humoral immune response in mice should facilitate studies to define the molecular basis for the transition from alloimmunity to autoimmunity in this model. We anticipate that characterization of monoclonal alloantibodies and autoantibodies in immunized mice may enable structural changes in antibody CDR to be identified that correlate with the postulated switch from alloimmunity to autoimmunity. Other, more powerful approaches can be used if this strategy proves unsatisfactory.52-54
Acknowledgments
This work was supported by grants HL-135285 (D.W.B.) and HL-13629 (R.H.A.) from the National Institutes of Health, National Heart Lung and Blood Institute. The studies were also supported by a generous gift from Rachel and Patrick English.
Authorship
Contribution: D.W.B. and R.H.A. conceived and designed the study, oversaw laboratory research activities, and wrote the manuscript; and D.W.B. and J.S. performed the laboratory studies.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Daniel W. Bougie, Blood Research Institute, versiti, PO Box 2178, Milwaukee, WI 53201; e-mail: dwbougie@versiti.org.

