

Key Points
The circRNAome is dramatically deregulated in T-ALL compared with normal thymocytes, with most circRNAs upregulated in malignant cells.
CircRNA-miRNA gene networks and circZNF609 silencing suggest a functional role for ectopically expressed circRNAs in T-ALL disease biology.

Abstract
Circular RNAs (circRNAs) are stable RNA molecules that can drive cancer through interactions with microRNAs and proteins and by the expression of circRNA encoded peptides. The aim of the study was to define the circRNA landscape and potential impact in T-cell acute lymphoblastic leukemia (T-ALL). Analysis by CirComPara of RNA-sequencing data from 25 T-ALL patients, immature, HOXA overexpressing, TLX1, TLX3, TAL1, or LMO2 rearranged, and from thymocyte populations of human healthy donors disclosed 68 554 circRNAs. Study of the top 3447 highly expressed circRNAs identified 944 circRNAs with significant differential expression between malignant T cells and normal counterparts, with most circRNAs displaying increased expression in T-ALL. Next, we defined subtype-specific circRNA signatures in molecular genetic subgroups of human T-ALL. In particular, circZNF609, circPSEN1, circKPNA5, and circCEP70 were upregulated in immature, circTASP1, circZBTB44, and circBACH1 in TLX3, circHACD1, and circSTAM in HOXA, circCAMSAP1 in TLX1, and circCASC15 in TAL-LMO. Backsplice sequences of 14 circRNAs ectopically expressed in T-ALL were confirmed, and overexpression of circRNAs in T-ALL with specific oncogenic lesions was substantiated by quantification in a panel of 13 human cell lines. An oncogenic role of circZNF609 in T-ALL was indicated by decreased cell viability upon silencing in vitro. Furthermore, functional predictions identified circRNA-microRNA gene axes informing modes of circRNA impact in molecular subtypes of human T-ALL.
Introduction
T-cell acute lymphoblastic leukemia (T-ALL) is a hematological malignancy caused by oncogenic transformation of developing thymocytes.1,2 Different genetic lesions have been recognized as T-ALL driving events, determining distinct genetic subgroups with specific gene,3,4 long noncoding RNA (lncRNA),5 and microRNA (miRNA)6 expression signatures. These molecular subtypes include TAL1/LMO2, TLX1, TLX3, NKX2.1, and HOXA rearranged T-ALLs. In addition, an immature/LYL1+ or early T-cell precursor ALL subgroup has been recognized with an early arrest during T-cell differentiation, often accompanied by myeloid marker expression.1,7
Circular RNAs (circRNAs) are transcripts in which a downstream splice donor site is covalently bound to an upstream acceptor site by a process called backsplicing, making these molecules particularly stable. The progressive discovery of functional roles of circRNAs and of their involvement in biological processes, with different mechanisms of action, made them attractive molecules for both fundamental and cancer research.8 By acting as miRNA sponges and competitive endogenous RNAs, circRNAs can indirectly regulate miRNA-target expression, ultimately controlling key miRNA-involving axes.9 Also, circRNAs can interact with RNA-binding proteins (RBPs)10 and regulate cellular processes.11 Beyond exerting functions typical of lncRNAs, circRNAs can be translated into peptides that are not encoded by linear transcripts.12-14 CircRNA-encoded peptides were shown to be biologically functional in the regulation of cell differentiation13 and cancer progression.15
Recent studies in normal hematopoiesis disclosed differential circRNA expression among blood cell types. Analysis of a set of circRNAs gave preliminary data on circRNA expression variation in different blood cell populations and maturation stages.16 Moreover, we provided detailed insight into circRNA expression of mature T cells.17
CircRNAs are implicated in hematopoietic malignancies, such as acute myeloid18 and MLL rearranged leukemias.19,20 However, a comprehensive analysis of circRNA expression in human T-ALL is still lacking.
Here, we used ribosomal-depleted RNA-sequencing (RNA-seq) to compare circRNA expression between human T-ALLs and normal T-cell progenitor cells. We identified different circRNA expression signatures in subtypes of human T-ALL. Analysis of circRNA, miRNA, and gene expression patterns in T-ALL highlighted putative oncogenic and tumor suppressor circRNAs, suggesting their functional involvement in the disease pathogenesis.
Methods
Patient and sample selection, cell sorting, and sequencing
Bone marrow lymphoblast samples from 25 T-ALL patients (5 for each of immature, TAL1/LMO2, TLX1, TLX3, and HOXA subtypes; supplemental Table 1) were collected with informed consent, according to the declaration of Helsinki from Saint-Louis Hospital, Paris, France. The study was approved by the Institut Universitaire d’Hematologie Institutional Review Board.21,22
Sorted human thymocyte populations were obtained from 2 healthy donors (supplemental Data). High-quality RNA samples from T-ALL cohort and from human thymocytes were used for high-depth Illumina total RNA sequencing (supplemental Data).
Bioinformatics analysis
CircRNAs were detected and quantified with CirComPara v0.6.323 combining the use of 6 backsplice detection methods (supplemental Data). CircRNAs reported by at least two of backsplice detection methods were considered detected; only circRNAs expressed in all samples of at least 1 T-ALL subgroup or T-cell maturation stage were further investigated (supplemental Data). CircRNA expression was normalized with the variance stabilizing transformation method.24 Differential expression was assessed by DESeq224 using Benjamini-Hochberg correction, considering significant differences with adjusted P ≤ .01. Circular to linear expression proportion (CLP) was computed as in Cheng et al25 (supplemental Data), and CircTest25 was used to assess significant CLP variation, representing a change of circRNA over the host gene expression, comparing T-ALL with thymocytes.
Different isoforms of interest were identified, specifying the backspliced exons or introns, using the longest Ensembl (GRCh38 and v93) transcript as reference.
Experimental validations and quantification in cell lines
CircRNA expression and backsplice junctions were confirmed using polymerase chain reaction (PCR) amplification with divergent primers in T-ALL cell lines (supplemental Table 2) and Sanger sequencing.
CircRNAs were quantified by quantitative reverse transcription (qRT)-PCR in T-ALL and non-T-ALL leukemia-derived cell lines (supplemental Data).
CircZNF609 silencing
To selectively silence circZNF609, we used small interfering RNA (siRNA)-circZNF609 described in Legnini et al13 and siRNA-NC26 as negative control. Cell transfection was performed in ALL-SIL and RPMI cell lines. At 24 and 48 hours from transfection and seeding, the level of circZNF609 was evaluated by Sybr green real-time polymerase chain reaction, and cell viability was tested by the MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) reduction assay. Detailed methods are in supplemental Data.
CircRNA functional predictions
MiRNA binding sites were predicted in circRNA sequences using miRanda,27 and strong validated miRNA target genes were retrieved with MIENTURNET28 (supplemental Data). Gene expression was quantified in the same samples considered for circRNA analysis, using StringTie v1.3.3. Cytoscape v3.7.2 was used to obtain circRNA-miRNA gene networks. RNA binding proteins recognition motifs were predicted by beRBP.29 The longest open reading frames overlapping the backsplice and continuing for at least 15 nt was considered (supplemental Data).
Results
CircRNAs expressed in T-ALL and developing thymocytes
To study the full spectrum of circRNAs expressed during malignant T-cell development, we analyzed high-depth ribodepleted RNA-seq data of 25 T-ALL patients representing 5 genetic T-ALL subtypes (immature, IMM; HOXA overexpressing, HOXA; TLX1 rearranged, TLX1; TLX3 rearranged, TLX3, and TAL1 or LMO2 rearranged, TAL-LMO; 5 patients per group; GSE110636). Five sorted human thymocyte populations from 2 healthy donors were used as normal T-cell counterparts: 3 CD34+ (CD34+4−1−, CD34+4−1+, CD34+4+) and 2 double positive (DP; CD4+CD8+CD3− and CD4+CD8+CD3+) populations. One donor’s CD34+ populations were sequenced twice (GSE142179) (Figure 1A; supplemental Table 1).
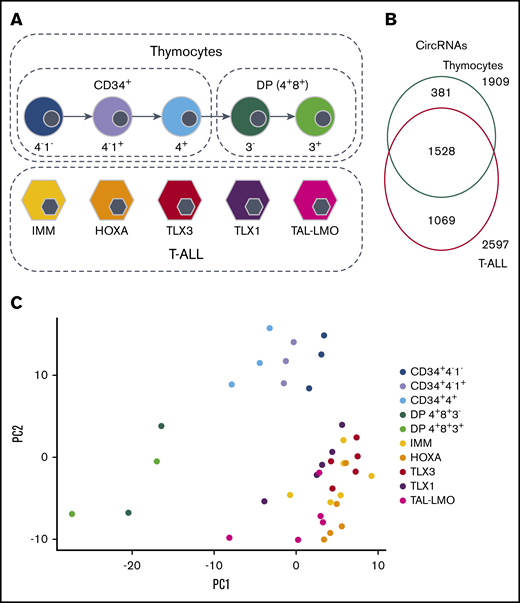
CircRNAs expression in 5 cytogenetic subtypes of T-ALL and normal thymocyte populations. (A) Study design and sample types. (B) Overlap of the 3447 circRNAs expressed in thymocytes and in T-ALL. (C) Samples displayed according to the 2 first principal components computed on the circRNA normalized expression.
CircRNAs expression in 5 cytogenetic subtypes of T-ALL and normal thymocyte populations. (A) Study design and sample types. (B) Overlap of the 3447 circRNAs expressed in thymocytes and in T-ALL. (C) Samples displayed according to the 2 first principal components computed on the circRNA normalized expression.
Quantification and annotation of circRNAs by CirComPara detected 68 554 circRNAs. After expression data normalization (supplemental Figure 1), we selected 3447 circRNAs that were highly expressed in all replicates of at least 1 sample group (see “Methods”), which derived from 1966 loci, mostly from exonic regions (89.4%). Among 8.3% of the circRNAs originated from introns, 1 circRNA (9: 111786793-111787947) derived from an intron of SHOC1. The large majority of the circRNAs (96.7%) derived from known loci of protein-coding genes, whereas only 1% generated from lncRNA genes. The remaining 2.3% of circRNAs corresponded to genomic regions with no genes annotated (intergenic circRNAs), and included 1 circRNA from chromosome X (X: 65051462-65075912) that was previously validated as highly expressed in lymphocytes and downregulated in B-cell acute lymphoblastic leukemia.17 Overall, 1264 (36.2%) circRNA host genes expressed multiple, up to 14, circular isoforms (supplemental Figure 2).
Examination of the circular to linear transcript expression proportion (CLP) gave a relative measure of each circRNA expression level compared with the linear transcripts overlapping the backsplice junctions. Supplemental Figure 3A-B shows the relation between the CLP and the absolute expression level of circRNAs in T-ALL and thymocytes. In particular, 297 (9%) circRNAs had a CLP ≥0.1, in T-ALL and/or in thymocytes, indicating that circRNA expression accounted for ∼10% of the total expression (linear plus circular) in the region spanned by the backsplice (supplemental Table 3). CircRNAs with the highest expression and/or CLP in T-ALL (Figure 2) included circSETD3 and circNEIL3, likely being the prevalent product of the host gene. Other highly expressed circRNAs, including circESYT2(9-13), had a relatively small CLP. The previously characterized circSETD3,17 circHIPK3,17,30 circFBXW7,17 and circZNF60915,17 were among the circRNAs with the highest expression and CLP in T-ALL. We identified circNBPF10, circLINS1, and circC9orf84, with particularly high CLP and accounting for most of the total expression of their host genes in T-ALL.
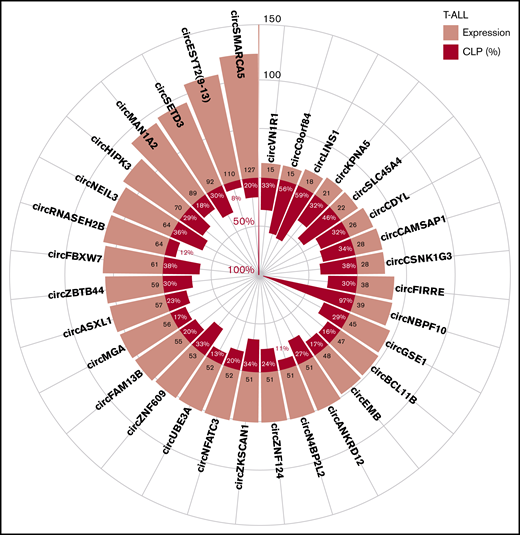
CircRNA with highest expression and/or circular to linear proportion in T-ALL. The plot shows the average absolute expression and the average circular to linear proportion (CLP) in T-ALL of the 32 circRNAs with average expression (normalized reads) >45 or with CLP > 0.3 and expression at least 10.
CircRNA with highest expression and/or circular to linear proportion in T-ALL. The plot shows the average absolute expression and the average circular to linear proportion (CLP) in T-ALL of the 32 circRNAs with average expression (normalized reads) >45 or with CLP > 0.3 and expression at least 10.
The circRNAome is deregulated in T-ALL
We investigated the expression difference between circRNAomes of human T-ALL cells and their normal T-cell progenitor counterparts, considering 1909 and 2597 circRNAs expressed in healthy thymocytes and T-ALLs, respectively. CircRNAs detected in both normal and malignant T cells were 54% (Figure 1B). Unsupervised principal component analysis of circRNA expression profiles (Figure 1C) separated T-ALL from thymocyte samples, pointing toward prominent differences in circRNA expression between normal and malignant T cells. In addition, according to circRNA expression, thymocyte populations tend to dispose in a gradient, from more immature to more mature. A slight separation of T-ALL molecular subtypes could also be noticed, even if overshadowed by the larger difference between normal and malignant cells.
Significantly different expression between T-ALLs and thymocytes was observed for 944 circRNAs (supplemental Table 4): 885 showed increased expression in leukemia, whereas only 59 were downregulated. Specifically, four were highly expressed in thymocytes and absent in malignant cells (Figure 3A), including circTFDP2 and an intergenic circRNA (circ15: 56480833-56492500, at 15q21.3). Furthermore, 5 circRNAs downregulated in T-ALL derived from a locus on chromosome 3p24.3, including SATB1-AS1 and SATB1, which encodes a protein essential for thymocyte maturation implicated in T-ALL pathogenesis.31
![CircRNA expression is deregulated in T-ALL. (A) Expression heatmap of the 103 circRNAs most deregulated in T-ALL compared with thymocytes from healthy donors (DESeq adjusted P < .01, log fold change [LFC] > 1, and mean expression >5; names are shown for the circRNAs with mean expression >10). (B) Scatterplot of significant absolute expression and CLP variation (LFC) comparing T-ALL with thymocytes; the marginal distributions of LFC(CLP) and LFC(Expression) values are shown, indicating the mean values; red and green dots indicate concordantly positive or negative LFC of expression and CLP, and dark yellow dots indicate discordant variations; names are shown for circRNAs with LFC(CLP) > LFC(Expression) and absolute expression of at least 10. Expr., expression.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/23/10.1182_bloodadvances.2020002337/2/m_advancesadv2020002337f3.png?Expires=1769080180&Signature=JwEFx7SFXmmtIPTsN04FCJCDlv8w660-3Qr7Pl6niciMyyGcurjDC6Pt08M5uFG63Zk1Evpu3hpCLZNQydQfsbN5tmPb-oMiL9QQcJWOy4cv4uHLXaoKMVeI2OvaVYc7Ulps0V7aPhBUsoxN5JWt6yf7~HuHcjtK0HpGbl1ykI8eApFDPTn2HgQi~DjGAJhas58B1ADzNGD1bu5m0BmX9cL00fyKE47eZzZ3QZVJa7R-GLGQcvE2H9vFy~Ggc~NDtbvR3-SQZ2C3S6YP5Q4q3as53FQhkYgT3GtKUor5C-j6idbnyUBzo4X0djuwv204JUs13NYzXqy7FZuGpH1-UA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CircRNA expression is deregulated in T-ALL. (A) Expression heatmap of the 103 circRNAs most deregulated in T-ALL compared with thymocytes from healthy donors (DESeq adjusted P < .01, log fold change [LFC] > 1, and mean expression >5; names are shown for the circRNAs with mean expression >10). (B) Scatterplot of significant absolute expression and CLP variation (LFC) comparing T-ALL with thymocytes; the marginal distributions of LFC(CLP) and LFC(Expression) values are shown, indicating the mean values; red and green dots indicate concordantly positive or negative LFC of expression and CLP, and dark yellow dots indicate discordant variations; names are shown for circRNAs with LFC(CLP) > LFC(Expression) and absolute expression of at least 10. Expr., expression.
CircRNA expression is deregulated in T-ALL. (A) Expression heatmap of the 103 circRNAs most deregulated in T-ALL compared with thymocytes from healthy donors (DESeq adjusted P < .01, log fold change [LFC] > 1, and mean expression >5; names are shown for the circRNAs with mean expression >10). (B) Scatterplot of significant absolute expression and CLP variation (LFC) comparing T-ALL with thymocytes; the marginal distributions of LFC(CLP) and LFC(Expression) values are shown, indicating the mean values; red and green dots indicate concordantly positive or negative LFC of expression and CLP, and dark yellow dots indicate discordant variations; names are shown for circRNAs with LFC(CLP) > LFC(Expression) and absolute expression of at least 10. Expr., expression.
The circRNAs most overexpressed in T-ALL (Figure 3A) included circRTN4, circPDE3B, circHACD1, circASXL1, circRBM23, circAC132807.2, circESYT2(4-8), circESYT2(9-13), and circMORC3. CircMORC3 was detected only in T-ALL samples of our RNA-seq data; expression in T-ALL cell lines was validated by qRT-PCR, and the backsplice junction was confirmed by Sanger sequencing (supplemental Table 2; supplemental Figure 4A-D).
The functional terms significantly enriched in the 810 genes corresponding to the 944 circRNAs deregulated in T-ALL are reported in supplemental Table 5. Several deregulated circRNAs derived from genes linked to NOTCH signaling, transcriptional regulation by p53 or SMAD2/SMAD3:SMAD4 heterotrimer and transforming growth factor-β (TGF-β) signaling, and from genes known to be upregulated in CD34+, CD8+ T cells, whole blood, and CD4+ T cells. CircRNAs that originated from other genes previously implicated in T-ALL disease biology were also deregulated. For instance, circZEB1 and 2 circLEF1 isoforms were downregulated, whereas circFBXW7 and circBCL11B were upregulated in T-ALL, with confirmed expression in T-ALL cell lines (supplemental Table 2; supplemental Figure 4B-D).
Most of the deregulated circRNAs (86%) derived from different genes. The 138 genes presenting multiple deregulated circRNAs showed predominantly concordant up- (126 genes; 97%) or downregulation (5) of all isoforms. The CASC15, PAN3, and UBAP2 genes showed several circRNAs that were upregulated in T-ALL, whereas multiple downregulated circRNAs derived from TFDP2, LEF1, and ZEB1. Only 7 genes had multiple circRNAs significantly differentially expressed in T-ALL with opposite behavior, including ANKRD36, which had 4 circular isoforms downregulated in malignant cells compared with thymocytes and two that were only expressed in T-ALL (supplemental Figure 5).
Next, we explored the expression variation of the circRNAs in relation to their linear counterparts considering the CLP variation, in T-ALL compared with normal T cells, conveying the rate of independence between a circRNA and the linear expression of the host gene.25 CircTest of significant CLP variations identified 133 circRNAs from 122 host genes with a significantly different CLP in T-ALL with respect to normal thymocytes. Thus, besides differences in absolute circRNA expression, several genes also display an imbalance of circular to linear transcripts proportion in T-ALL as compared with normal thymic precursors. Matching the circRNA with significant differential absolute expression and CLP between T-ALL and thymocytes revealed that most of the absolute expression variations of circRNAs did not correspond to a varied CLP, indicating that circRNA expression followed the variation of the linear counterpart. Of note, a CLP variation was observed for 54 differentially expressed circRNAs (Figure 3B), mostly (47/54) concordant with expression changes. Nevertheless, for 23 circRNAs, the CLP increase in T-ALL was more pronounced when compared with the increase of the absolute expression. Thus, overexpression in T-ALL of these circRNAs, including the highly expressed circBCL11B, circKDM1A(2-9), circNFATC3(2), and circZSCAN1, was not linked to a corresponding increase of the linear counterpart. CircBCL11B absolute expression was increased in T-ALL (LFC 0.55), less strikingly than its CLP (LFC 1.28), indicating that the circBCL11B proportion over the linear transcript of BCL11B gene is deregulated in malignant T cells.
CircRNAs with T-ALL group-specific ectopic expression: predicted functions and interactions
Previous studies have shown that genetic subtypes of human T-ALL are characterized by unique messenger RNA, miRNA, and lncRNA expression signatures. Principal component analysis based on circRNAs expression in T-ALL revealed that these molecular genetic entities also display specific circRNA expression profiles (supplemental Figure 6). Comparing each molecular genetic subtype vs the others, 86 circRNAs were significantly differentially expressed (supplemental Figure 7; supplemental Table 4). Unsupervised analysis based on these 86 subtype-specific circRNAs revealed that IMM and TLX3 samples cluster together, whereas a distinct group was composed of TAL-LMO, TLX1, and HOXA leukemias (Figure 4A; supplemental Figure 7), with a clear discrimination of TLX1 and TLX3 samples, in line with Wallaert at al.6 Expression of most of the 86 circRNAs differentially expressed in T-ALL molecular subtypes followed the linear counterpart. Only 13 circRNAs had CLPs that significantly vary among groups, including 4 circRNAs for which the CLP varied in the T-ALL subtype that also showed circRNA expression deregulation. Of note, both circYPEL1 expression and CLP were decreased in TLX1, indicating uncoupled expression of circRNA and its linear counterpart.
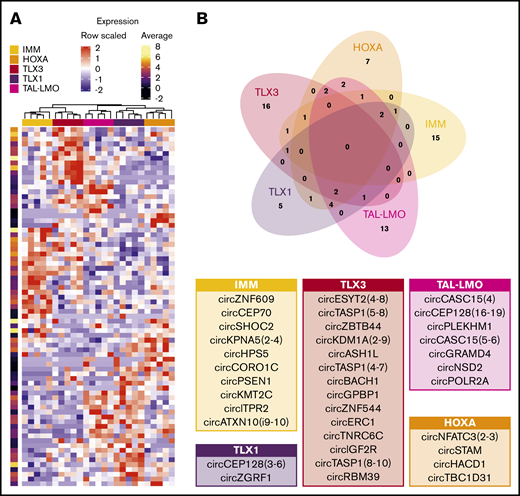
Specificities of circRNAs expression in T-ALL subgroups. (A) Expression profiles of the 75 circRNAs upregulated in at least 1 T-ALL group (DESeq adjusted P < .01; standardized expression values and per row log average expression are shown in the heatmap and in the bar on the right, respectively). (B) Venn diagram of circRNAs upregulated in each T-ALL subgroup overlap defines the circRNAs upregulated with group specificity; tables indicate circRNAs with average expression in the group >10 showing in larger characters those with expression >30.
Specificities of circRNAs expression in T-ALL subgroups. (A) Expression profiles of the 75 circRNAs upregulated in at least 1 T-ALL group (DESeq adjusted P < .01; standardized expression values and per row log average expression are shown in the heatmap and in the bar on the right, respectively). (B) Venn diagram of circRNAs upregulated in each T-ALL subgroup overlap defines the circRNAs upregulated with group specificity; tables indicate circRNAs with average expression in the group >10 showing in larger characters those with expression >30.
Notably, 56 circRNAs were upregulated in specific groups of T-ALL: 15 in IMM, 7 in HOXA, 16 in TLX3, 5 in TLX1, and 13 in TAL-LMO (Figure 4B). For most (48; 85.7%) of these circRNAs, expression did not vary when comparing normal CD34+ and DP thymocytes. For the remaining 8 circRNAs, the expression differences between T-ALL subtypes might reflect the varying expression during thymocyte maturation of corresponding counterparts.
With the aim to identify circRNAs with significantly increased (ectopic) expression in a specific T-ALL group compared with normal counterparts, T-ALL subtypes were compared with normal thymocytes, and to CD34+ and DP thymocytes separately. Fourteen circRNAs with ectopic expression in specific groups of T-ALL were identified (supplemental Table 4). CircZNF609, circCEP70, circKPNA5, and circPSEN1 were ectopically expressed in IMM, whereas circESYT2(4-8), circKDM1A(2-9), circTASP1(4-7), circTASP1(5-8), circZBTB4, and circBACH1 were ectopically expressed in TLX3. In HOXA, the ectopic expression of circSTAM and circHACD1 was defined. CircCAMSAP1 and circCASC15 were ectopically expressed in the more mature TLX1 and TAL-LMO T-ALL subtypes, respectively. We validated 11 circRNAs with ectopic upregulation in T-ALL subgroups, compared with thymocytes, and confirmed the backsplice junctions by Sanger sequencing (supplemental Table 2; Figure 5).
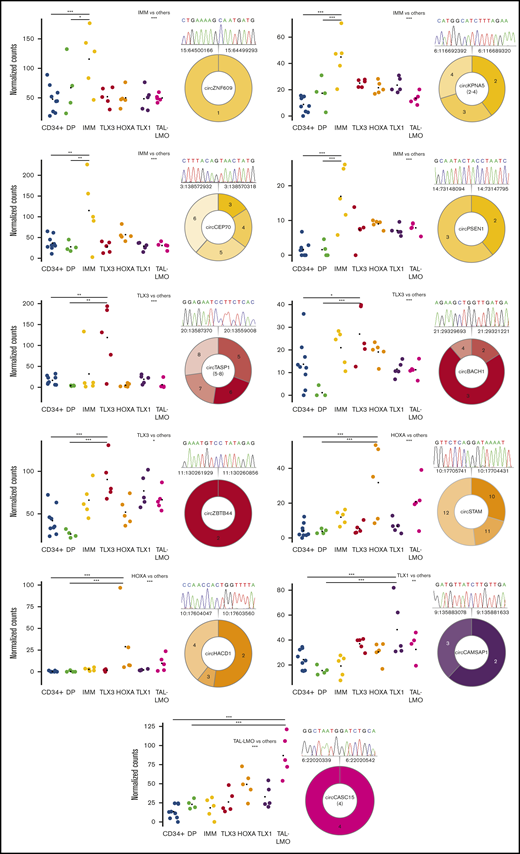
Prominent circRNAs upregulated with T-ALL group specificity. Expression in CD34+ and in DP developing thymocytes from healthy donors and in samples of different T-ALL groups of 11 circRNAs significantly upregulated in 1 T-ALL group compared with all other T-ALL samples, with CD34+ and with DP thymocytes (***, **, and * indicate differential expression with adjusted P < .001, <.01, and <.05, respectively). The predicted exon structure with the backsplice junction validated by RT-PCR and Sanger sequencing is shown for each circRNA (different colors indicate the group specificity and different color shades exons putatively included in the circRNA; the chromatogram depicts the sequence of the backsplice junction exactly corresponding to that identified by RNA-seq).
Prominent circRNAs upregulated with T-ALL group specificity. Expression in CD34+ and in DP developing thymocytes from healthy donors and in samples of different T-ALL groups of 11 circRNAs significantly upregulated in 1 T-ALL group compared with all other T-ALL samples, with CD34+ and with DP thymocytes (***, **, and * indicate differential expression with adjusted P < .001, <.01, and <.05, respectively). The predicted exon structure with the backsplice junction validated by RT-PCR and Sanger sequencing is shown for each circRNA (different colors indicate the group specificity and different color shades exons putatively included in the circRNA; the chromatogram depicts the sequence of the backsplice junction exactly corresponding to that identified by RNA-seq).
Furthermore, RT-PCR quantification of 14 circRNAs in a panel of 13 cell lines showed a clear tendency toward higher expression in ALL cell lines compared with acute myeloid leukemia (AML) in most cases (Figure 6; supplemental Figure 4D). Distinctive expression in T-ALL lines was observed for circBCL11B, circCASC15, circTASP1, and circKPNA5. Considering circRNA expression levels in relation to the molecular subtypes circTASP1 and circZBTB44, high expression in the DND-41 cell line of the TLX3 oncogenic subgroup was in line with their overexpression in TLX3 T-ALL patients. Also, circCASC15 and circCAMSAP1 overexpressed in TLX1 and in TAL-LMO T-ALL, respectively, had high, although not specific, expression in cell lines assigned to these molecular groups.
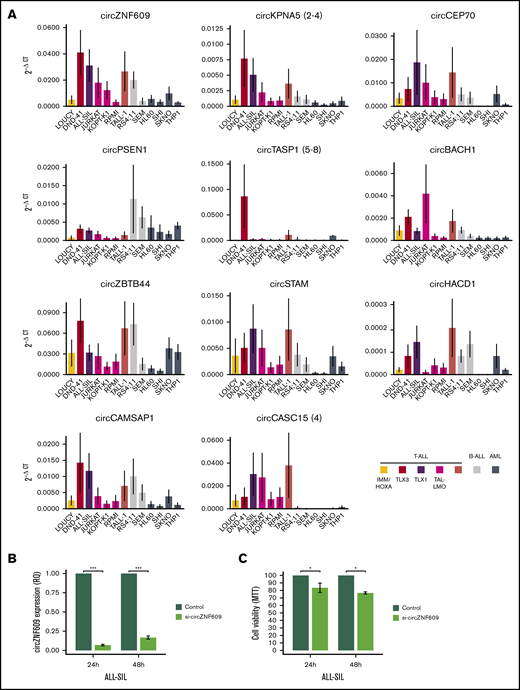
CircRNA expression in acute leukemia cell lines and decreased cell viability upon circZNF609 silencing in T-ALL in vitro. (A) CircRNA expression quantification by qRT-PCR in a panel of 13 acute leukemia cell lines of the T-cell, B-cell, and myeloid lineages (mean ± standard error of 2−ΔCT in 4 replicates per cell line). As indicated in the legend, T-ALL, B-ALL, AML cell lines are shown in different colors, and the oncogenic group for T-ALL cell lines is specified, when available (human T-ALL cell lines database: https://humantallcelllines.wordpress.com). (B) Efficient circZNF609 silencing was obtained in the ALL-SIL cell line upon transfection with a specific siRNA (mean ± standard error of relative quantification in 3 independent experiments). (C) Low proliferation and cell viability (metabolic proliferation assay) in ALL-SIL cells with reduced expression of circZNF609 compared with control, at 24 and 48 hours after transfection and seeding (mean ± standard error of 3 independent experiments). ***, **, and * indicate P < .001, <.01, and <.05, respectively (Mann-Whitney U test). B-ALL, B-cell acute lymphoblastic leukemia.
CircRNA expression in acute leukemia cell lines and decreased cell viability upon circZNF609 silencing in T-ALL in vitro. (A) CircRNA expression quantification by qRT-PCR in a panel of 13 acute leukemia cell lines of the T-cell, B-cell, and myeloid lineages (mean ± standard error of 2−ΔCT in 4 replicates per cell line). As indicated in the legend, T-ALL, B-ALL, AML cell lines are shown in different colors, and the oncogenic group for T-ALL cell lines is specified, when available (human T-ALL cell lines database: https://humantallcelllines.wordpress.com). (B) Efficient circZNF609 silencing was obtained in the ALL-SIL cell line upon transfection with a specific siRNA (mean ± standard error of relative quantification in 3 independent experiments). (C) Low proliferation and cell viability (metabolic proliferation assay) in ALL-SIL cells with reduced expression of circZNF609 compared with control, at 24 and 48 hours after transfection and seeding (mean ± standard error of 3 independent experiments). ***, **, and * indicate P < .001, <.01, and <.05, respectively (Mann-Whitney U test). B-ALL, B-cell acute lymphoblastic leukemia.
The functional role of circZNF609 has been previously described, showing that it favors proliferation of immature myoblasts and plays an oncogenic role in embryonal rhabdomyosarcoma.15 CircZNF609 upregulation in T-ALL with high absolute and relative expression motivated functional investigation also in T-ALL. An efficient silencing of circZNF609 was obtained in the ALL-SIL (Figure 6B) and RPMI T-ALL cell lines (supplemental Figure 8A). Of note, significant decrease of cell viability at 24 and 48 hours after circZNF609 was observed in ALL-SIL cells (Figure 6C). Differently, circZNF609 silencing did not affect RPMI cell viability (supplemental Figure 8B). A considerably higher baseline expression of circZNF609 in ALL-SIL than in RPMI cells (Figure 6A) possibly explains this observation.
To further explore molecular mechanisms of ectopic upregulated circRNAs in T-ALL subgroups, functional predictions were highly instrumental. RBP binding motifs were identified (supplemental Table 6), and RBPs with multiple binding sites included SRSF1, EIF4B, LIN28B, and CSDA. In addition, 3 circRNAs with an open reading frame containing the backsplice sequence potentially translating into circRNA-specific peptides were detected by analysis of coding potential (supplemental Table 7).
Next, for each T-ALL molecular subtype, a circRNA-miRNA gene interaction network was obtained, linking validated circRNAs with ectopic upregulation in the T-ALL group to miRNAs previously associated with and having a tumor suppressor role in T-ALL development.2,6,32 In turn, miRNAs were connected to validated target genes upregulated in samples of the same T-ALL molecular subtype (supplemental Figures 9-10). For instance, the IMM T-ALL regulatory network included 4 circRNAs and 21 genes upregulated in IMM, connected through 15 miRNAs (supplemental Figure 9). Interactions between circRNAs and miRNAs previously associated with the same molecular subtype6 were outlined, with circZNF609 and miR-181 both linked to IMM, circZBTB44, and miR-let-7i-5p to TLX3, circHACD1, and miR-182-5p to HOXA. Taken together, the connections of circRNAs upregulated in T-ALL subtypes with miRNAs (Figure 7) indicated a plausible convergence of multiple deregulated circRNAs to the same miRNAs, most notably miR-150, miR-34, and miR-181.
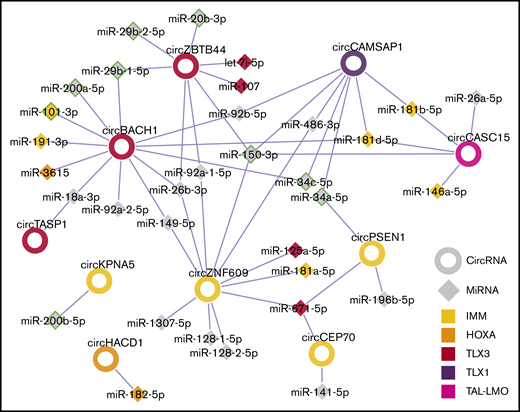
Summary of putative circRNAs interactions with miRNAs in T-ALL molecular subtypes. The network shows circRNA (rings) upregulated in T-ALL molecular subtypes linked to miRNAs (diamonds), according to miRNA recognition element predictions in circRNA sequences. CircRNA and miRNA node fill color indicates the molecular subtype identified, respectively, in this study and in Wallaert et al.6 A green border indicates miRNAs with tumor suppressor role in T-ALL.2,32
Summary of putative circRNAs interactions with miRNAs in T-ALL molecular subtypes. The network shows circRNA (rings) upregulated in T-ALL molecular subtypes linked to miRNAs (diamonds), according to miRNA recognition element predictions in circRNA sequences. CircRNA and miRNA node fill color indicates the molecular subtype identified, respectively, in this study and in Wallaert et al.6 A green border indicates miRNAs with tumor suppressor role in T-ALL.2,32
Discussion
In this study, we analyzed circRNA expression of 25 T-ALL samples, comprising 5 genetic subtypes, and normal thymocyte populations. Reliable circRNA detection from RNA-seq data was obtained by 6 different backsplice detection methods,33 integrated in CirComPara.23 Stringent selection criteria identified 3447 circRNAs that were explored for sample group comparisons and to extract the most informative circRNAs.
As a first highlight, we observed that the T-ALL circRNAome was dramatically deregulated, as witnessed by the high number of differentially expressed circRNAs, that were mainly upregulated in T-ALLs compared with normal thymic counterparts. The majority (96%) of differentially expressed circRNAs were common for the T-ALL samples, hinting at a shared mechanism of circRNA deregulation, irrespective of the acquired genetic background.
The dysregulation of the T-ALL circRNAome can be ascribed to epigenetic and transcriptional alterations by activated oncogenic pathways and to aberrant splicing, in line with data of splicing factor deregulation in T-ALL.34 Although we identified a number of circRNAs that were the prevalent transcript of their host gene, the expression of most circRNAs in T-ALL followed the pattern of the corresponding linear transcript. Interestingly, several overexpressed circRNAs derived from genes associated with T-ALL oncogenic signaling, such as NOTCH135-37 (circFBXW7, circCDK8, and circRAB6A), PI3K/AKT, TGF-β,38 and TP53.39
Of the circRNAs deregulated in T-ALL, 34 were previously described as part of a set of 102 circRNAs showing variable expression during hematopoiesis,16 including circNFATC3(2), circSPECC1, circANKRD12, circCCDC66, circESYT2(9-13), and circCDK13. In addition, a role in cancer was proven for circBACH140 and circHIPK3,17,41 and in our analysis was specifically upregulated in T-ALL with differences among subgroups. Heterogeneous circFBXW7 expression in T-ALL patients is intriguing for the role of FBXW7 in the NOTCH1 pathway and for the recently shown increased proliferation of AML cells upon circFBXW7 knockdown,42 indicating circFBXW7 as a tumor suppressor.
Relatively few circRNAs were less expressed in malignant cells, and further study is needed to identify a potential tumor suppressor role in T-ALL. Conversely, circRNAs overexpressed in T-ALL can include onco-circRNAs. Notably, for circRNAs detected herein, molecular functions and mechanisms pointing at oncogenic potential were previously described in other contexts. CircUBAP2 was shown to be upregulated in triple-negative breast cancer, where it sponges miRNA-661 counteracting the repression of the MTA1 oncogene,43 required for BCL11B transcriptional repression activity in T cells.44 Increase in T-ALL of the pro-proliferative circHIPK3 is in line with its overexpression in various solid cancers and in B-cell precursor ALL.17 CircHIPK3 could favor derepression of TAL1 and c-MYBs through suppression of several miRNAs, particularly miR-124,30 that targets TAL1 and c-MYBs.45 CircRTN4(2-3) was recently demonstrated to be translated,46 but the role of the encoded peptide is still unknown. Most of circRNAs overexpressed in T-ALL are not yet functionally characterized, including several expressed from leukemia-associated genes, such as circASXL1 and the validated circMORC3 and circBCL11B. Our data showed that circBCL11B is also highly expressed in T-ALL cell lines. The significant increase of circBCL11B circular to linear proportion, when comparing T-ALL with thymocytes (Figure 3B), opens an intriguing hypothesis about regulation of BCL11B, a key regulator of the initial stages of human T-cell differentiation47 that is frequently mutated or deleted in T-ALL.
The second highlight of this study is the identification of circRNAs expressed with group specificity and possibly playing oncogenic roles in distinct T-ALL subtypes. Of particular interest were circRNAs, whose expression in a T-ALL group is ectopic or significantly exceeding the levels observed in the closest normal counterpart.
CircCAMSAP1 was upregulated in TLX1; it had high CLP (0.42), and it was expressed less in the IMM group than in more mature subtypes. CircCAMSAP1’s oncogenic role in solid cancer48 encourages investigating this circRNA also in T-ALL. Our predictions linked circCAMSAP1 to 7 miRNAs, including miR-150 and miR-34 with tumor suppressor role in T-ALL.
The IMM group was characterized by 4 circRNAs overexpressed with group specificity and the absence of variation during thymocyte maturation: circZNF609, circPSEN1, circCEP70, and circKPNA5. CircZNF609 upregulation in IMM T-ALL was particularly intriguing considering the known oncogenic potential of this circRNA12-14 and the previously detected upregulation of linear ZNF609 transcripts in IMM5 that, according to the high circZNF609 CLP observed, can be partially due to the overexpression of the circRNA. Our in vitro experiments indicated that circZNF609 sustains cell proliferation in T-ALL, which is in line with recent data from solid tumors. CircZNF609’s oncogenic features have been linked both to the encoded peptide15 and to a miRNA-sponging activity.49 Bioinformatics functional predictions and laboratory testing that prioritize circZNF609 will experimentally establish disease-related mechanisms in T-ALL.
Network analysis suggests new regulatory axes by linking the circRNAs ectopically expressed in IMM T-ALL to miRNAs and to validated target genes that are upregulated. Several genes of the MAPK signaling pathway (MAP2K1, AKT1, FASLG, ARRB2, FOS, and EPHA2) were involved. MiR-34a, a recognized tumor suppressor in T cell,32 is putatively sponged by circPSEN1. CircZNF609 was linked to miR-181a-5p, characteristic of the IMM group,6 and to miR-125a-5p. Both of these miRNAs target LNFG, which boosts the leukemogenic potential of NOTCH1 signaling in T-ALL.50
The immature TLX3 group was characterized by overexpression of circESYT2(4-8), circKDM1A(2-9), circTASP1(4-7), circTASP1(5-8), circZBTB44, and circBACH1. For the last three with decreased expression during thymocyte maturation, the upregulation in TLX3 T-ALL was significant compared with immature thymocytes. These circRNAs were also highly expressed in T-ALL cell lines. Patient and cell line data concordantly indicated high circTASP1(5-8) and circZBTB44 expression linked to the TLX3 oncogenic subtype. CircBACH1 was recently shown to promote hepatocellular carcinoma growth40 by repressing p27 translation. The regulatory network involving circTASP1(5-8), circZBTB44, and circBACH1, ectopically expressed in TLX3 T-ALL, included 7 tumor suppressor miRNAs plus miR-107 and let-7i-5p, which were previously associated with the TLX3 molecular subtype.6 The validated target genes upregulated in TLX3 included SMAD3, a key factor and therapeutic target in T-ALL51,52 ; NAP1L1, a MLLT10 translocation partner in T-ALL53 with antiapoptotic activity by enhancement of NF-κB signaling pathway54 ; ENO1, a metabolic gene55 ; SPIRE1, an actin organizer; CERS2, a gene conferring chemoresistance in T-ALL56 ; and ABHD17C, which affects Ras localization and function.
CircSTAM and circHACD1, ectopically expressed in HOXA, were not previously characterized. A possible circHACD1 function was identified of derepressing miR-182-5p targets previously linked to HOXA T-ALL, including HOXA9, a main driver of leukemogenesis,57 and other oncogenes, such as THBS1.58
TAL-LMO T-ALL was characterized by high expression of circCASC15 isoforms, derived from a known RNA oncogene (CASC15/LncSox4), which were upregulated in the same T-ALL group.5 CircCASC15(4) was highly expressed only in T-ALL cell lines. CircCASC15(4) has been proposed in bladder cancer as a therapeutic target promoting cell proliferation.59 Our data suggested a possible link of circCASC15(4) with 5 miRNAs and 18 upregulated genes, including the leukemia-associated genes FGFR1, IQCG, KCTD15, and S100A12.
Mounting evidence is linking circRNA regulatory activity to interaction with RBPs,60 such as circFOX3, that affects cell proliferation and apoptosis through binding to p21 and mdm2, respectively. In this light, functional hypotheses for circRNAs associated with T-ALL are elicited by the predicted interactions with RBPs. Splicing factors, such as the prooncoprotein SRSF1 whose knock-down increases apoptosis of leukemic cells,61 and EIF4B factor with proleukemic activity62 are appealing.
In summary, this study provided new evidence of circRNA deregulation in T-ALL and of circRNA expression variation in distinct T-ALL subtypes, extending previous data on linear transcripts expression and informing circRNAs dysregulation from genes hitherto not associated with specific T-ALL groups. Upregulated circRNAs for which oncogenic functions have been proven in different settings or that are still not characterized are attractive candidates for functional studies in T-ALL. Our experimental data about the pro-proliferative role of circZNF609 and the predicted regulatory circRNA-miRNA target gene networks generated new hypotheses of disease mechanisms warranting further investigation.
RNA-seq data have been deposited in Gene Expression Omnibus database (accession number GSE110636).
Acknowledgments
This work has been supported by Italian Ministry of Education, Universities, and Research grant PRIN 2017 2017PPS2X4_003 (S.B. and G.t.K.), AIRC, Milan, Italy Investigator Grant 2017 20052 (S.B.), MFAG 2018 21771 (V.S.), Fondazione Umberto Veronesi, Milan, Italy (E.G.), Fund for Scientific Research Flanders and Concerted Research Action (GOA) of the Ghent University (P.V.V., F.S., and T.T.), ERC St Grant Consolidator 311660 (S.G.), and the ANR-10-IBHU-0002 Saint-Louis Institute program (S.G.).
Authorship
Contribution: A.B., M.P., G.t.K., P.V.V., and S.B. conceived the study; A.B., E.G., A.D.M., and S.B. contributed bioinformatics methods and data analysis; M.P., G.G., and M.T.S. performed circRNA experimental validations and quantification in cell lines; V.S. and M.P. performed circZNF609 silencing experiments; J.R., M.D.D., K.D., S.G., F.S., T.T., and P.V.V. provided RNA-seq data; A.B. and S.B. drafted the manuscript; A.B., M.P., and S.B. made the figures; and all authors revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Stefania Bortoluzzi, Department of Molecular Medicine, University of Padua, Via G. Colombo 3, 35131 Padua, Italy; e-mail: stefania.bortoluzzi@unipd.it; and Maddalena Paganin, Onco-Hematology, Stem Cell Transplant and Gene Therapy Laboratory, Fondazione Città della Speranza Istituto di Ricerca Pediatrica, Corso Stati Uniti 4, 35127 Padua, Italy; e-mail: maddalena.paganin@unipd.it.

