

Key Points
CD30 CAR T-cell therapy promoted a prolonged remission in a patient with multiply relapsed EATL.
Introduction
Enteropathy-associated T-cell lymphoma (EATL) is a rare lymphoma arising in the setting of celiac disease.1 Multiagent anthracycline-based chemotherapy alone is associated with poor long-term outcomes with a median overall survival of only 7 months.2,3 In patients who achieve a response to first-line therapy and are eligible for autologous stem cell transplantation (SCT), 5-year median overall survival can be improved to 50% to 60%.3-5 Because ∼50% of patients with EATL express the CD30 antigen, targeted therapy with brentuximab vedotin has been explored with promising outcomes.6,7 We have recently completed a phase 1/2 study showing safety and efficacy of chimeric antigen receptor (CAR)–modified T cells targeting the CD30 molecule (CD30 CAR T cells) in CD30+ Hodgkin lymphoma.8 Here we describe a patient with multiply relapsed EATL after previous allogeneic SCT (allo-SCT) who achieved a durable remission with CD30 CAR T cells.
Case description
A 69-year-old male with a history of celiac disease, which was well-controlled with a gluten-free diet for 9 years, developed abdominal pain and nausea. He underwent endoscopy and biopsy, which revealed CD30+ EATL of the small bowel. A positron emission tomography/computed tomography (PET/CT) scan at diagnosis demonstrated extensive hypermetabolic lymphadenopathy above and below the diaphragm and extranodal disease in the small bowel. The patient was refractory to cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) and ifosfamide, carboplatin and etoposide (ICE), but he achieved a partial response (PR) after 3 cycles of brentuximab vedotin and bendamustine and received consolidation therapy with a nonmyeloablative allo-SCT from a matched related donor. He received maintenance brentuximab vedotin after allo-SCT, but treatment was discontinued after 5 cycles because of neuropathy.
Ten months after his allo-SCT, he developed a new cutaneous nodule, and a biopsy confirmed relapsed EATL. He achieved a complete response (CR) with local radiation and brentuximab vedotin but became refractory to brentuximab vedotin after 5 months. He subsequently was treated with 2 cycles of romidepsin and donor lymphocyte infusion (DLI; 1.46 × 107 cells per kg) and achieved a short-lived PR (<3 months). He received 2 cycles of brentuximab vedotin and a second DLI (1.0 × 108 cells per kg) resulting in a brief CR lasting <2 months. A new biopsy of a cervical lymph node confirmed relapsed CD30+ EATL (Figure 1A-B). Given that his disease was refractory to chemotherapy and brentuximab vedotin, he was referred to the University of North Carolina (UNC) for treatment with CD30 CAR T cells.
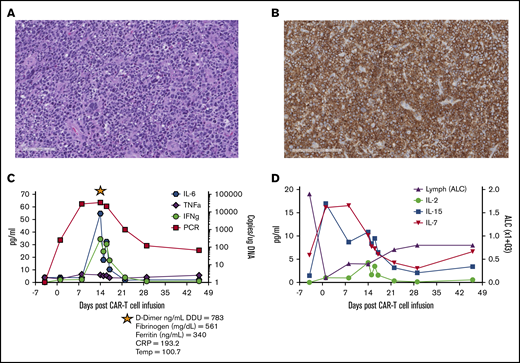
Pre–CAR-T diagnostic biopsy, CD30CAR-T expansion, and detection of homeostatic cytokines. Hematoxylin and eosin stain (A; scale bar, 100 μm) and CD30 antibody stain of the tumor biopsy before CAR T-cell therapy (B; scale bar, 200 μm). (C) Detection of CD30 CAR T-cell molecular signals by quantitative polymerase chain reaction (qPCR) and of the indicated cytokines over the course of the first 6 weeks after infusion. The star indicates evaluation of the listed parameters during the CRS event. (D) Detection of IL-2, IL-7, and IL-15 in the plasma and lymphocyte counts over the course of the first 6 weeks after infusion. ALC, absolute lymphocyte count; CRP, C-reactive protein; DDU, D-dimer unit; IFNg, interferon-γ; TNFa, tumor necrosis factor-α.
Pre–CAR-T diagnostic biopsy, CD30CAR-T expansion, and detection of homeostatic cytokines. Hematoxylin and eosin stain (A; scale bar, 100 μm) and CD30 antibody stain of the tumor biopsy before CAR T-cell therapy (B; scale bar, 200 μm). (C) Detection of CD30 CAR T-cell molecular signals by quantitative polymerase chain reaction (qPCR) and of the indicated cytokines over the course of the first 6 weeks after infusion. The star indicates evaluation of the listed parameters during the CRS event. (D) Detection of IL-2, IL-7, and IL-15 in the plasma and lymphocyte counts over the course of the first 6 weeks after infusion. ALC, absolute lymphocyte count; CRP, C-reactive protein; DDU, D-dimer unit; IFNg, interferon-γ; TNFa, tumor necrosis factor-α.
Methods
This patient was treated on our phase 1b Institutional Review Board–approved clinical trial (NCT02690545). Lymphodepleting chemotherapy consisted of bendamustine 70 mg/m2 per day and fludarabine 30 mg/m2 per day followed by 2 × 108 CD30 CAR T cells per m2, manufactured at the UNC Good Manufacturing Practice–compliant facility (IND14688).8 Disease response was determined by PET/CT imaging at 6 weeks after CAR T-cell therapy using Lugano criteria.9 Cytokine release syndrome (CRS) was graded according to Lee’s criteria.10 Persistence of CD30 CAR T cells in vivo was determined by quantitative polymerase chain reaction that used peripheral blood samples.8 Cytokines were measured in the plasma by Luminex assay.
Results and discussion
The CD30 CAR T-cell product characteristics were similar to those described for CAR T cells generated from patients with Hodgkin lymphoma.8 Specifically, after 20 days of ex vivo culture, the product was composed of 99% T cells that expressed the CD30 CAR, which contained both CD8+ (48%) and CD4+ (50%) cells, with phenotypic characteristics of effector-memory T cells (CD45RA–CCR7–; 75%) and a modest population of central-memory T cells (CD45RA–CCR7+; 10%). Of note, he maintained 100% donor T-cell chimerism before CAR T-cell production, making it likely that CAR T cells were generated from the allograft. He received 3 days of lymphodepleting chemotherapy followed by CD30 CAR T cells at a dose of 2 × 108 CAR T cells per m2.8
Treatment with lymphodepletion and infusion of CD30 CAR T cells was well tolerated, with grade 3 or higher toxicities limited to lymphopenia during the first 6 weeks, which was most likely related to lymphodepleting chemotherapy. The patient developed grade 1 CRS on day 12, which corresponded to the peak of CAR T-cell expansion and increased inflammatory cytokines, such as interleukin-6 (IL-6) and interferon-γ in the peripheral blood (Figure 1C). Other CRS-related parameters included elevated levels of C-reactive protein, ferritin, fibrinogen, and D-dimer. CRS resolved spontaneously by day 15 and did not require specific therapy. Although expansion of CD30 CAR T cells in this patient was associated with an increase in the homeostatic cytokines IL-7 and IL-15 after lymphodepletion (Figure 1D), CD30 CAR T cells were no longer detectable by quantitative polymerase chain reaction 3 months after infusion.8
Disease evaluation by PET/CT before receiving lymphodepletion and CD30 CAR T cells showed involvement of bilateral cervical, supraclavicular, and mediastinal nodes as well as a single mesenteric node. All locations completely resolved 6 weeks after CD30 CAR T-cell therapy (Figure 2). Routine imaging at 24 months revealed continued remission, and the patient remains clinically well on follow-up physical examinations more than 30 months after treatment.
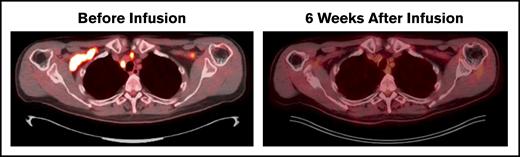
Durable CR after CD30 CAR T-cell therapy. Patient with multiply relapsed EATL. Before lymphodepletion and CD30 CAR T-cell infusion, disease involved the bilateral cervical, supraclavicular, mediastinal, hilar, and a single mesenteric lymph node. Six weeks after infusion, the patient achieved a Deauville score of 1 and CR. Additional disease assessments at 6, 12, and 24 months have continued to show CR.
Durable CR after CD30 CAR T-cell therapy. Patient with multiply relapsed EATL. Before lymphodepletion and CD30 CAR T-cell infusion, disease involved the bilateral cervical, supraclavicular, mediastinal, hilar, and a single mesenteric lymph node. Six weeks after infusion, the patient achieved a Deauville score of 1 and CR. Additional disease assessments at 6, 12, and 24 months have continued to show CR.
There is limited experience in manufacturing autologous CAR T cells from individuals with T-cell lymphomas (TCLs). Here we show the feasibility of achieving therapeutic doses of CD30 CAR T cells in a patient with EATL. To our knowledge, this is the first case of EATL treated with CAR T-cell therapy. In this patient, who had not achieved a durable response with 6 previous lines of therapy, including brentuximab vedotin, allo-SCT, and multiple DLIs, we are encouraged by both the depth and duration of response after CD30 CAR T-cell therapy. Given his previous history of allo-SCT and 100% donor chimerism at the time of CAR T-cell production, we suspect that the CD30 CAR T cells were produced from the allograft. The impact of previous allo-SCT on the success of CD30 CAR T-cell treatment in this patient is not known, although this should be investigated in future studies.
There is very limited experience with CD30 CAR T-cell therapy in treating patients with CD30+ TCLs. The patient described in this report was the only patient with EATL treated with CD30 CAR T cells on our clinical trial. An additional adult with CD30+ Sézary syndrome was treated with the same lymphodepletion and cell dose, achieved a PR at 6 weeks with reduction in his modified Severity Weighted Assessment Tool (mSWAT) score from 26 to 1, but developed a clinical relapse after 9 weeks of follow-up. Ramos et al8 reported phase 1 dose escalation results of CD30 CAR T cells without lymphodepletion chemotherapy, which included 2 patients with anaplastic large cell lymphoma (ALCL). The first patient, who had cutaneous anaplastic lymphoma kinase–negative ALCL, received a dose of 2 × 107 CAR T cells per m2 (one-log lower than our patient) with no response. The second patient with systemic anaplastic lymphoma kinase–positive ALCL achieved a durable CR (9 months) after receiving 4 separate CD30 CAR T-cell infusions at a dose of 2 × 108 CAR T cells per m2.11 A second group, evaluating a slightly different CD30 CAR construct, reported 1 patient with cutaneous ALCL treated with CD30 CAR T cells at a dose of 1.5 × 107 cells per kg without previous lymphodepletion who obtained a PR for 3 months.12
Although only ALCL and certain subsets of TCLs express CD30, targeting this molecule with a CAR still offers numerous advantages. First, fratricide is minimal, which guarantees that there will be no deleterious effects during T-cell product manufacturing. This is in contrast to CAR T cells targeting other T-cell lineage–associated antigens, such as CD7, CD5, and CD4.13-15 Second, CD30 CAR T cells have a very tolerable safety profile compared with toxicities anticipated with CAR T cells targeting other T-cell lineage antigens. CD30 CAR T cells have no reported deleterious effects on cellular immunity, suggesting that the risk for opportunistic infections should be minimal. In addition, episodes of CRS seem less severe compared with those in which CAR T cells target other antigens such as CD19. Finally, neurotoxicity has not been observed in patients receiving CD30 CAR T-cell therapy.
Well-designed clinical trials are needed to expand our understanding of and overcome obstacles when using CD30 CAR T cells in TCL. The remarkable response observed in this patient should be confirmed in a larger phase 2 trial. On the basis of experience with CD19 CAR expansion and persistence, CAR T cells can be used as surrogate markers for clinical outcomes. It will be important to develop methods for increasing expansion and persistence of the infused CD30 CAR T cells and therefore improve clinical outcomes. In addition, each distinct TCL subset may present different obstacles to the persistence of CAR T cells, possibly related to the tumor environment. Finally, any effect of CD30 antigen density on clinical responses should be investigated. To this end, UNC is currently enrolling patients with relapsed/refractory CD30+ peripheral TCL in a phase 2 trial that uses 2 sequential CD30 CAR T-cell infusions at the recommended phase 2 dose of 2 × 108 CAR T cells per m2 (NCT04083495).
Presented in abstract form at the 12th Annual T-Cell Lymphoma Forum, La Jolla, CA, 31 January 2020.
Acknowledgments
This work was supported by a grant from the National Heart, Lung, and Blood Institute, National Institutes of Health (RO1HL114564) (B.S.), by the University Cancer Research Fund at the Lineberger Comprehensive Cancer Center (B.S. and G.D.), and by a grant from Stand Up To Cancer (7000000853) (G.D.).
Authorship
Contribution: T.J.V., N. Grover, B.S., and A.W.B. conducted the research and wrote the manuscript; A.W.B., N. Ghosh, J.B., and C.C. participated in the patient’s care; T.J.V., G.D., B.S., N. Grover, and J.S. discussed and interpreted the results; and K.M., A.I., and J.S. wrote the clinical protocol.
Conflict-of-interest disclosure: Competing interests of authors from the University of North Carolina are managed in accordance with institutional policies. B.S., G.D., and J.S. have a pending patent concerning the CD30 CAR-T product presented in this article. J.S. has filed an intellectual property patent regarding the use of STING agonists to enhance CAR T-cell therapy. N. Grover served as a consultant for Tessa Therapeutics, has received research funding from Genentech, and has served on an advisory board for Kite Pharma. T.J.V. received research funding from AstraZeneca. The University of North Carolina has a research collaboration with Tessa Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Timothy J. Voorhees, Lineberger Comprehensive Cancer Center, University of North Carolina–Chapel Hill, 170 Manning Dr, CB 7305, Chapel Hill, NC 27599-7305; e-mail: timothy.voorhees@unchealth.unc.edu.

