

Key Points
The incidence of aGVHD grade 2-4 after αβ T-cell depletion is 26% at day 100, allowing prophylactic DLI in the majority of patients.
The incidence of moderate and severe cGVHD at 2 years is 17% and 0%, respectively, comparing favorably to T-cell–replete allo-HSCT.

Abstract
We conducted a multicenter prospective single-arm phase 1/2 study that assesses the outcome of αβ T-cell depleted allogeneic hematopoietic stem cell transplantation (allo-HSCT) of peripheral blood derived stem cells from matched related, or unrelated donors (10/10 and 9/10) in adults, with the incidence of acute graft-versus-host disease (aGVHD) as the primary end point at day 100. Thirty-five adults (median age, 59; range, 19-69 years) were enrolled. Conditioning consisted of antithymocyte globulin, busulfan, and fludarabine, followed by 28 days of mycophenolic acid after allo-HSCT. The minimal follow-up time was 24 months. The median number of infused CD34+ cells and αβ T cells were 6.1 × 106 and 16.3 × 103 cells per kg, respectively. The cumulative incidence (CI) of aGVHD grades 2-4 and 3-4 at day 100 was 26% and 14%. One secondary graft failure was observed. A prophylactic donor lymphocyte infusion (DLI) (1 × 105 CD3+ T cells per kg) was administered to 54% of the subjects, resulting in a CI of aGVHD grades 2-4 and 3-4 to 37% and 17% at 2 years. Immune monitoring revealed an early reconstitution of natural killer (NK) and γδ T cells. Cytomegalovirus reactivation associated with expansion of memory-like NK cells. The CI of relapse was 29%, and the nonrelapse mortality 32% at 2 years. The 2-year CI of chronic GVHD (cGVHD) was 23%, of which 17% was moderate. We conclude that only 26% of patients developed aGVHD 2-4 after αβ T-cell–depleted allo-HSCT within 100 days and was associated with a low incidence of cGVHD after 2 years. This trial was registered at www.trialregister.nl as #NL4767.
Introduction
αβ T cells have a crucial role in the pathology of acute graft-versus-host disease (aGVHD), especially shortly after allogeneic hematopoietic stem cell transplantation (allo-HSCT), when inflammation is induced by chemotherapy conditioning of the recipient.1,2 Therefore, major developments in transplantation strategies include alternative dosing of αβ T cells, during and after allo-HSCT. High-dose cyclophosphamide has been shown to preferentially target proliferating, alloreactive T cells3 and is increasingly used as posttransplantation immune prophylaxis with cyclophosphamide (PTCY).4-7 Alternatively, a stringent in vivo and ex vivo T-cell depletion has long been known as an effective strategy to prevent severe GVHD.8 Antithymocyte globulin (ATG) is widely used to deplete T cells in vivo.9 Also, use of alemtuzumab has a long history as a T-cell depletion strategy. A variety of approaches have been reported, such as in vivo and “in vitro-in the bag” T-cell depletion methods, with no consensus or standardization of this method.10 More recently, both CD34 selection11-13 and CD3 and αβTCR/CD19 depletion have entered clinical practice.14-17
αβ T-cell depletion of grafts before allo-HSCT has been pioneered in pediatric patients with both malignant and nonmalignant diseases when using a haploidentical donor.14,15,17-19 Because the reported incidences of aGVHD and chronic GVHD (cGVHD) appeared to be very favorable compared with what has been reported in T-cell–repleted allo-HSCT, we proposed that αβ T-cell–depleted allo-HSCT could also be a promising low-toxicity allo-HSCT platform for patients with malignant diseases, with a matched related or unrelated donor (MRD/MUD).
Here, we present the prospective analysis of an adult cohort, which received an αβ T-cell–depleted stem cell product of peripheral blood derived stem cells (PBSCs), derived from MRD/MUD. Conditioning started with early ATG to minimize ATG exposure at the day of infusion.20 Subsequently, a myeloablative reduced toxicity conditioning regimen consisting of busulfan and fludarabine was administered. We report the incidence of aGVHD at day 100, which is the primary outcome of this study. In addition, we analyzed secondary clinical endpoints at the 2-year follow-up.
Patients and methods
Clinical cohort
Adults with a range of hematological malignancies were enrolled between 29 April 2016 and 12 October 2017 in this multicenter prospective clinical trial, approved by the local ethical committee, and registered at the Dutch trial registry. Written informed consent was obtained in accordance with the Helsinki Declaration.
Eligibility, donor selection, and donor treatment are reported in the supplemental Methods.
Study treatment
The conditioning regimen consisted of ATG (Thymoglobulin) 1.5 mg/kg IV days −12 to −9, fludarabine IV 40 mg/m2 days −5 to −2, and busulfan IV (Busilvex) days −5 to −2. The ATG dosage prediction model was based on a modified pediatric population pharmacokinetic/pharmacodynamic model, in which body weight and average lymphocyte counts at baseline were used as variables.20,21 The ATG dosage was aimed at a post-HSCT area under the curve of ∼20 to 30 AU*day/mL. Therapeutic drug monitoring of busulfan was performed and targeted to a cumulative area under the curve of 80 to 90 mg/h per L.22 αβ T-cell reduction was performed by depletion with anti-αβ T-cell receptor (TCR) antibodies, in combination with magnetic microbeads using the automated CliniMACS device (Miltenyi Biotec, Bergisch Gladbach, Germany), as described previously,23 and according to standard operating procedures. The maximal allowed residual αβ T-cell number was 5 × 105 cells per kg. The allograft was infused within 24 hours after depletion. Mycophenolate mofetil (MMF) was administered at a dose of 15 mg/kg, 3 times a day (maximum, 3000 mg total) for 28 days as a single GVHD prophylaxis. Prevention and treatment strategies of infections are reported in the supplemental Methods. After completion of the study protocol, patients received a preemptive donor lymphocyte infusion (DLI) (1 × 105 CD3+ T cells per kg) at 3 months after stem cell infusion, in line with guidelines for T-cell depletion, provided patients were without GVHD, and without immunosuppressive medication at the time of DLI.2
Clinical definitions
Definitions of neutrophil and platelet recovery, as well as primary and secondary graft failures are reported in the supplemental Methods. All time-to-event end points were measured from the date of stem cell infusion. Patients were evaluated systematically for aGVHD and cGVHD. aGVHD was staged and graded according to the Glucksberg classification (updated according to Przepiorka et al24 ). cGVHD was staged according to the National Institute of Health consensus criteria.25 Cumulative incidence (CI) of aGVHD was defined as time to onset of GVHD, with relapse and death as competing events. Overall survival (OS) was defined as time to death from any cause. The CI of relapse is defined as time to relapse, with death as a competing event. Nonrelapse mortality (NRM) was defined as time to death, without relapse or progression. Event-free survival (EFS) was defined as the time to relapse/disease progression, graft failure, or death, whichever came first. CI of cytomegalovirus (CMV) and Epstein-Barr virus (EBV) reactivations were calculated with relapse and death as competing events.
Flow cytometry analysis and functional analysis NK cell subsets
PBMCs were prospectively collected at 3 months and 6 months in most eligible patients. As a control population for natural killer (NK) cell analysis either samples of PBMCs stored in the Biobank HSCT (protocol METC 11-063) (patient details in supplemental Table 1) or PBMCs collected from healthy blood donors were used. Frozen PBMCs were thawed and rested overnight. For phenotypical analysis, cells were stained with fluorochrome-conjugated antibodies detailed in supplemental Table 2. The gating strategy to dissect NK cells subsets is depicted in supplemental Figure 1. Cells were acquired by either a Fortessa or Canto flow cytometer and analyzed by DIVA software. For functional analysis, frozen PBMCs were thawed and rested overnight and (where indicated) stimulated with recombinant interleukin-12 (IL-12; 10 ng/mL) and IL-18 (100 ng/mL). Cells were permeabilized with 0.1% Triton X-100 and interferon-γ production was evaluated by flow cytometry.
Statistical analysis
OS and EFS were calculated by Kaplan-Meier estimates and were expressed as percentage and 95% confidence interval (type log-log) using R package “survival” (version 2.44.1.1). Loss to follow-up was censored. CI probabilities with competing risks were estimated using R package cmprsk (version 2.2.9), with the function “CumIncidence()” as described elsewhere.26 Calculations are based on Fine & Gray competing risk models.26-28 Reconstitution of immune subsets was summarized by Prism, version 8, software (GraphPad). Differences were analyzed using the Mann-Whitney U test.
Results
The clinical cohort, graft composition, and engraftment
Thirty-five patients with a broad range of hematological malignancies including acute myeloid leukemia (AML)/myelodysplastic syndrome with excess of blasts-2 (40%), multiple myeloma (MM) (14%), and myeloproliferative neoplasm (11%) were included in this prospective study (Table 1). The median age was 59 years (range, 19-69 years). Donors were MRD (11%), 10/10 MUD (60%), and a high percentage of 9/10 MUD donors (29%). αβ T-cell depletion was successfully performed for all allografts, resulting in a median 4.1 log depletion of αβ T cells (data not shown). The median number of infused CD34+ cells and αβ T cells were 6.1 × 106 cells per kg (range, 2.8-10) and 0.02 × 106 cells per kg (range, 0.00-0.09), respectively (Figure 1). Median numbers of infused γδ T cells were 5.1 × 106 cells per kg (range, 0.5-36.5), median numbers of NK cells were 18.4 × 106 cells per kg (range, 8.7-59.6), and median number of B cells were 41.3 × 106 cells per kg (range, 12.6-88.6). All patients alive at the time of analysis had a minimal follow-up of 24 months. The control T-cell–replete reduced intensity conditioning cohort used for the analyses of NK cell subsets is described in supplemental Table 1.
Clinical characteristics
| Characteristic | Value, n (%) |
|---|---|
| Median age (range), y | 59 (19-69) |
| Female | 10 (29) |
| Median time post-allo-HSCT (range), mo | 35 (24-42) |
| Disease | |
| AML/MDS-EB2 | 13 (37) |
| ALL | 4 (11) |
| CML | 2 (6) |
| MM | 5 (14) |
| NHL | 1 (3) |
| MPN | 4 (11) |
| CMML | 2 (6) |
| MDS* | 4 (11) |
| Donor type | |
| MRD | 4 (11) |
| MUD (10/10) | 21 (60) |
| MMUD (9/10) | 10 (29) |
| DLI (1 × 105 CD3 cells per kg) | 19 (54) |
| Median time to DLI (range), wk | 11 (7-16) |
| CMV recipient/donor | |
| +/+ | 9 (26) |
| +/− | 8 (23) |
| −/+ | 4 (11) |
| −/− | 14 (40) |
| EBV recipient/donor | |
| +/+ | 23 (66) |
| +/− | 4 (11) |
| −/+ | 4 (11) |
| −/− | 0 (0) |
| +/unknown | 4 (11) |
| Characteristic | Value, n (%) |
|---|---|
| Median age (range), y | 59 (19-69) |
| Female | 10 (29) |
| Median time post-allo-HSCT (range), mo | 35 (24-42) |
| Disease | |
| AML/MDS-EB2 | 13 (37) |
| ALL | 4 (11) |
| CML | 2 (6) |
| MM | 5 (14) |
| NHL | 1 (3) |
| MPN | 4 (11) |
| CMML | 2 (6) |
| MDS* | 4 (11) |
| Donor type | |
| MRD | 4 (11) |
| MUD (10/10) | 21 (60) |
| MMUD (9/10) | 10 (29) |
| DLI (1 × 105 CD3 cells per kg) | 19 (54) |
| Median time to DLI (range), wk | 11 (7-16) |
| CMV recipient/donor | |
| +/+ | 9 (26) |
| +/− | 8 (23) |
| −/+ | 4 (11) |
| −/− | 14 (40) |
| EBV recipient/donor | |
| +/+ | 23 (66) |
| +/− | 4 (11) |
| −/+ | 4 (11) |
| −/− | 0 (0) |
| +/unknown | 4 (11) |
CML, chronic myeloid lymphoma; CMML, chronic myelomonocytic leukemia; MDS-EB2, myelodysplastic syndrome with excess of blasts-2; MPN, myeloproliferative neoplasm; NHL, non-Hodgkin lymphoma.
*Indicates all MDS except MDS-EB2.
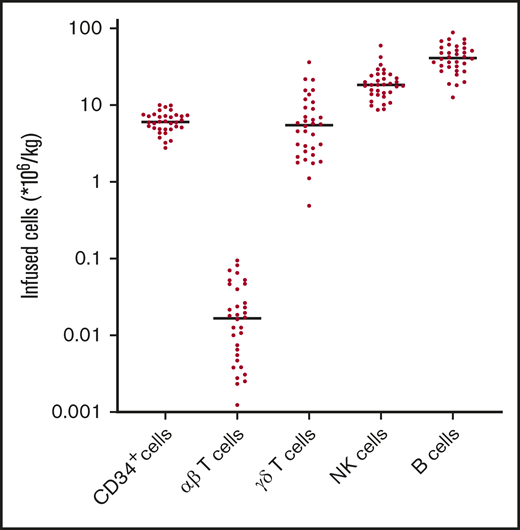
Graft composition after αβ T-cell depletion. Total number of CD34+ cells, αβ T cells, γδ T cells, NK cells, and B cells in the infused allograft were measured by flow cytometric analysis. Bullets represent individual numbers; dash represents the median.
Graft composition after αβ T-cell depletion. Total number of CD34+ cells, αβ T cells, γδ T cells, NK cells, and B cells in the infused allograft were measured by flow cytometric analysis. Bullets represent individual numbers; dash represents the median.
Engraftment and GVHD
The median time to neutrophil engraftment was 14 days (range, 9-48 days), and the median time to platelet engraftment was 17 days (range, 10-99 days) (supplemental Table 3). One secondary graft failure (day 56) was observed, potentially because of an early CMV reactivation (data not shown). The CI of aGVHD grade 2-4 and grade 3-4 at day 100 was 26% (95% confidence interval, 13-41) and 14% (5-28), respectively (Figure 2A). A prophylactic DLI derived from frozen leftover material of the mobilized stem cell product, containing 1 × 105 CD3+ T cells per kg was administered to 54% of the patients after a median of 11 weeks (Table 1). aGVHD grade 1-4 (N = 13) was the major reason for not administering a prophylactic DLI. Other reasons included NRM <3 M (n = 1), graft failure (n = 1), and relapse <3 M (n = 1). After 2 years, the CI of aGVHD grade 2-4 and grade 3-4 was increased to 37% (21%-53%) and 17% (7%-31%), respectively (Figure 2A), likely as consequences of the prophylactic DLI. The CI of cGVHD at 2 years was 23% (1%-38%), of which 17.1% (6.7%-31.6%) of total subjects were classified moderate and none of the subjects were classified severe (Figure 2B).
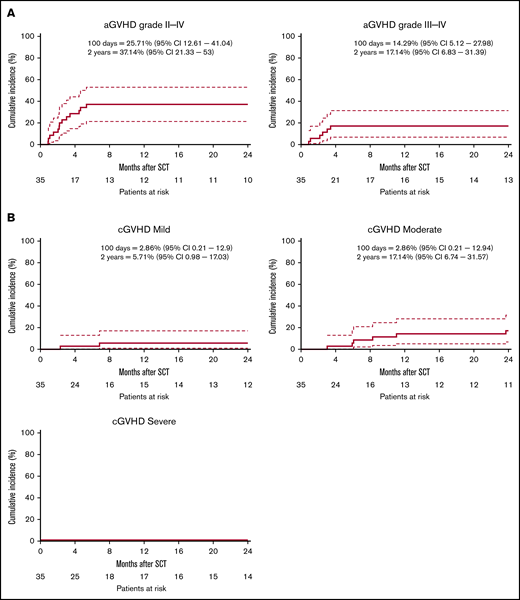
Cumulative incidence aGVHD and cGVHD. The cumulative incidence (CI) of GVHD is defined as time to onset of GVHD, with relapse and death as competing events. (A) CI of aGVHD ≥grade 2-4 (left) and aGVHD ≥3-4 (right). (B) CI of mild cGVHD (left), moderate GVHD (right), or severe cGVHD (bottom left). Dashed lines indicate a 95% confidence interval (95% CI).
Cumulative incidence aGVHD and cGVHD. The cumulative incidence (CI) of GVHD is defined as time to onset of GVHD, with relapse and death as competing events. (A) CI of aGVHD ≥grade 2-4 (left) and aGVHD ≥3-4 (right). (B) CI of mild cGVHD (left), moderate GVHD (right), or severe cGVHD (bottom left). Dashed lines indicate a 95% confidence interval (95% CI).
Viral infections and immune reconstitution
CMV and EBV infections are major drivers of severe infectious complications after allo-HSCT. Therefore, viral loads were captured systematically during the study period. The CI of CMV reactivation in patients at risk (CMV seropositive recipients) was 41% (18%-63%) at 100 days and 65% (36%-83%) at 2 years (Figure 3). One of the patients developed CMV disease. All other patients were successfully treated preemptively. The CI of CMV in the complete study cohort was 34% (19%-50.1%) at 2 years (supplemental Figure 2). The CI of EBV reactivation in the complete cohort was 32% (17%-48%) at 100 days and 44% (27%-60%) at 2 years (Figure 3). One patient presented with posttransplant lymphoproliferative disorder. All other reactivations were successfully treated with rituximab. Analysis of all infectious complications within the first year of allo-HSCT revealed that the majority of patients did not experience serious infectious complications (defined as Common Terminology Criteria for Adverse Events grades 3-5) and that most serious infections could be successfully treated (supplemental Table 4).
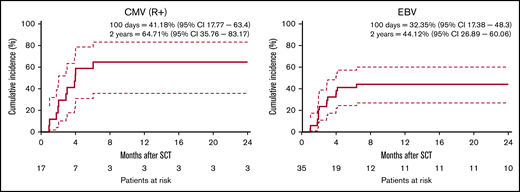
Cumulative incidence of viral reactions. Cumulative incidence of CMV and EBV infections are calculated with relapse and death as competing events. (A) CI of CMV reactivation for patients at risk (defined as seropositive recipients, R+). (B) CI of EBV reactivation. Dashed lines indicate 95% CI.
Cumulative incidence of viral reactions. Cumulative incidence of CMV and EBV infections are calculated with relapse and death as competing events. (A) CI of CMV reactivation for patients at risk (defined as seropositive recipients, R+). (B) CI of EBV reactivation. Dashed lines indicate 95% CI.
Given the severe αβ T-cell depletion, we next addressed the level of immune reconstitution (IR) after allo-HSCT. In line with our assumption that sparing NK cells and γδ T cells would allow for a rapid reconstitution of these innate immune subsets, NK cell IR was swift, with a median number of NK cells of 182 NK cells per μL at day 100, which remained stable over time (Figure 4). In line with previous observation after αβTCR/CD19-depleted haplo-SCT,17 CMV reactivation resulted in a significant increase in memory-like NK cells (defined as CD56+CD57+NKG2C+),29 both at 3 and 6 months (Figure 5). This increase was not observed in a T-cell–replete control cohort. The percentage of NKG2A−/KIR+ cells was not affected (supplemental Figure 3). As expected,17 however, this did not result in an increase in interferon-γ production after stimulation with IL-12/IL-18 (Figure 5).
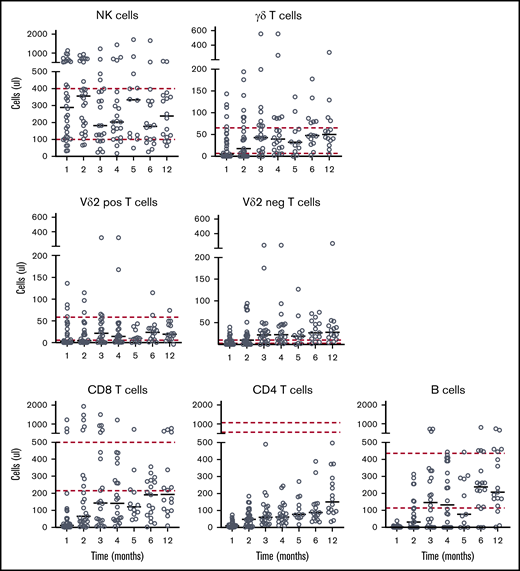
Immune reconstitution. Peripheral blood samples were collected at the indicated time intervals after allo-HSCT. Absolute numbers of indicated immune subsets were measured by flow cytometric analysis. Circles indicate individual values; lines indicate the median value; dashed lines represent the normal range in healthy individuals.
Immune reconstitution. Peripheral blood samples were collected at the indicated time intervals after allo-HSCT. Absolute numbers of indicated immune subsets were measured by flow cytometric analysis. Circles indicate individual values; lines indicate the median value; dashed lines represent the normal range in healthy individuals.
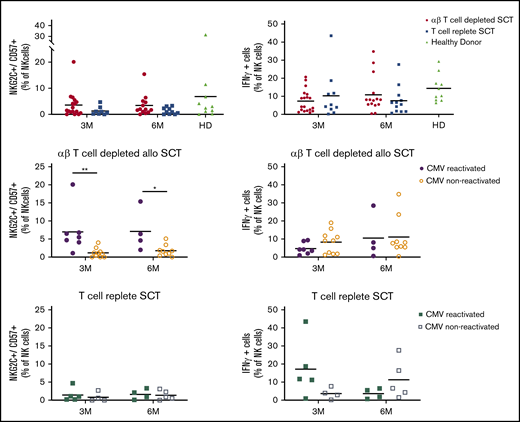
NK cell analysis. NK cell repertoire at 3 and 6 months after allo-SCT. Where indicated, transplant samples were stratified for CMV-reactivated recipients (filled symbols) or CMV-seronegative recipients (open symbols). Bullets depict recipients of the prospective αβ T-cell–depleted cohort; squares depict recipients of the T-cell–replete cohort; triangles depict healthy donors. All cell fractions are presented as fraction of NK cells. Statistics: Mann-Whitney U test (*P < .05; **P < .01). Comparisons without indication of significance where not statistically different.
NK cell analysis. NK cell repertoire at 3 and 6 months after allo-SCT. Where indicated, transplant samples were stratified for CMV-reactivated recipients (filled symbols) or CMV-seronegative recipients (open symbols). Bullets depict recipients of the prospective αβ T-cell–depleted cohort; squares depict recipients of the T-cell–replete cohort; triangles depict healthy donors. All cell fractions are presented as fraction of NK cells. Statistics: Mann-Whitney U test (*P < .05; **P < .01). Comparisons without indication of significance where not statistically different.
Also, a rapid γδ T-cell IR was observed in the majority of patients. γδ T-cell IR included both Vδ2+ γδ T cells, as well as Vδ2− γδ T cells. As expected, γδ T-cell numbers were significantly lower compared with NK cells (median numbers of total γδT cells 43 cells per μL, Vδ2+ γδ T 21 cells per μL, and Vδ2− γδ T cells 22 cells per μL at day 100; Figure 4). The median number of CD8+ αβ T cells were 143 cells per μL at day 100. With median numbers of CD4+ αβ T cells of 60 cells per μL at day 100, CD4+ αβ T cells reconstituted more slowly. This is in line with reported data for other transplantation regimens.30 Despite only a selective αβ T-cell depletion being performed, median B-cell IR was also impaired during the first months in a large fraction of patients, probably because the CI of EBV was 44, requiring the administration of rituximab. Nevertheless, immunoglobulin values normalized in 58% percent of the patients at month 12 (supplemental Figure 4). Interestingly, though heterogeneous, we observed that patients receiving DLI had a trend toward an improved immune reconstitution in terms of T, B, and NK cells most of the time prior administration of DLI (supplemental Figure 5).
Disease control and survival
Despite a rather diverse composition of diseases, with different reported OS after allo-HSCT,31 we formally assessed the OS of the total study cohort as the secondary end point. At 2 years after treatment, the OS within the total cohort was 54% (37%-67%) and the EFS was 40% (24%-56%) (Figure 6). The CI of relapse was 29% (15%-44%) at and the NRM was 32% (17%-48%) at 2 years. Analysis of the cause of death showed that toxicity (both GVHD and infections) was the major contributor to mortality within 2 years of follow-up, whereas relapse accounted for the minority of deaths (supplemental Figure 6). Within patients that died because of NRM (n = 12), either GVHD alone (n = 3) or GVHD with subsequent (respiratory) infections (n = 4) were the major causes leading to death. Of note, patients with MM (n = 5) had a very poor EFS (supplemental Figure 7) because of early NRM or progression.
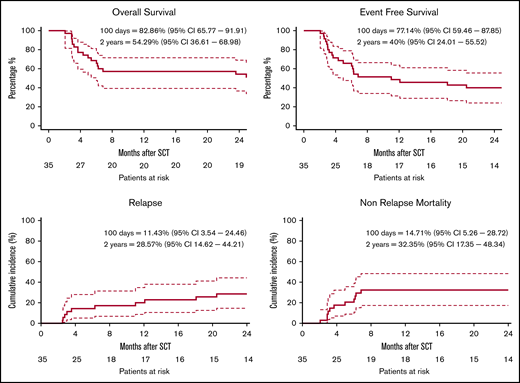
Clinical outcome. Clinical outcomes are shown with a 2-year follow-up. OS (A) and EFS (B) were estimated by the Kaplan-Meier product. (C) The CI of relapse is defined as time to relapse, with death as a competing event. (D) NRM is defined as time to death, without relapse or progression. Dashed lines indicate 95% CI.
Clinical outcome. Clinical outcomes are shown with a 2-year follow-up. OS (A) and EFS (B) were estimated by the Kaplan-Meier product. (C) The CI of relapse is defined as time to relapse, with death as a competing event. (D) NRM is defined as time to death, without relapse or progression. Dashed lines indicate 95% CI.
Discussion
We conducted the first prospective multicenter study using a selective αβ T-cell depletion strategy in adults with hematological malignancies. We observed a low incidence of grade 2-4 aGVHD at day 100 (26%), with immune suppression limited to 4 weeks MMF after transplant, rendering the majority of patients eligible for early postalloimmune interventions. Consequently, the incidence of aGVHD grade 2-4 increased to 37% at 2 years and was somewhat higher when compared with aGVHD after selective CD34+ selection that was between 16% to 23% at 2 years.12,13 In contrast for PTCY, the Johns Hopkins Group reports an incidence of grade 2-4 aGVHD 40% to 60% after 1 year in a study where bone marrow was used as a stem cell source. More recently, studies analyzing PBSCs as a stem cell source, and PTCY in combination with tacrolimus and MMF as a GVHD prophylaxis reported an incidence of grade 2-4 GVHD of ∼30%.32 Also, adding ATG to transplantation regimens can result in comparable incidences of aGVHD,33,34 particularly when dosed appropriately. NRM was in our cohort 32% at 2 years at a first sight higher when compared with 14% to 17% as reported for PTCY5,35 and 21% to 24% reported for CD34-selected allo-SCT.12,13 Given the high confidence intervals of our study because of rather low patient numbers as compared with other reports, these data are difficult to interpret. A high incidental amount of MM patients with all fatal outcomes in our cohort might partially account for these differences.
In the complete cohort, the incidence of cGVHD remained low. In fact, it was comparable to what has been observed when bone marrow was used in combination with PTCY for haploidentical donors (7%-21%),6 or 9/10 MUD donors (9%-28%)34 or when compared with CD34 selection (5%-19%)12,13 and substantially reduced when compared with T-cell–replete therapy with MRD and MUD (overall ∼50%).36 Because most allo-HSCT platforms, including PTCY, require a prolonged immune suppression vastly exceeding the 4 weeks used in our study, it remains challenging to administer early postalloimmune interventions to the majority of patients with most other regimens. DLI can be administered prophylactically or preemptively to prevent early relapse by promoting IR.37 In our study, we were able to administer a prophylactic DLI to more than 50% of the patients. As such, our data emphasize the potential power of our reported αβ T-cell depletion platform. Interestingly, patients receiving a DLI after αβ T-cell depletion had already an improved IR when compared with patients not eligible for DLI. This suggests that a balanced IR in total numbers favors a clinical status that allows to administer DLIs and that the additive value of a DLI arises from enriching the TCR repertoire diversity rather than an increase in total numbers.38
Sufficient control of infections is the second important denominator of a low-toxicity HSCT platform. The CI of CMV in our cohort was 34% in the total cohort and 64% in the patients at risk. This is both in line with T-cell–replete allo-HSCT,36,39,40 as well as after PTCY and ATG,41,42 which have incidences of CMV reactivations of around 30% in complete patient populations as well as with CD34 selection, which reports a comparable incidence of CMV reactivations of 70% in seropositive patients.43 Thus, control of CMV appears to be comparable within most HSCT platforms; however, mechanisms of IR most likely differ substantially. Because of the low number of patients, we could not assess whether an early DLI could further decrease the number of CMV infections. Because most CMV infections occurred before day 100, it is unlikely that a DLI substantially contributed to a better CMV control. A well-balanced early IR might be more important. In a αβ T-cell depletion platform, early control of infections most likely relies substantially on NK and γδ T cells. These subsets do not require prolonged immune suppression, because neither NK nor γδ T cells44,45 mediate alloreactivity. After CD34+ selection, early reconstitution of NK cells only is apparently also sufficient to control CMV reactivation,46 though no data are available on viral loads to compare potencies of different immune subsets. Our observation that CMV reactivation associates with an expansion of memory-like NK cells emphasizes the role of NK cells also in αβ T-cell–depleted cohorts, as also reported by others.47 In contrast, patients receiving PTCY are substantially impaired in NK cells, and most likely are more competent in αβ T cells, which also explains the need for the prolonged immune suppression used within the context of the strategy.48
EBV reactivation was observed in 44% of the recipients, which is consistent with the results of other T-cell depletion strategies like alemtuzumab, which had a reported incidence of 48% in a retrospective analysis.49 This is surprising, given that γ9δ2T cells have been reported to sufficiently recognize EBV transformed LCL cell lines.50 However, for the mode of action, administration of a bisphosphonate, such as pamidronate, is hypothesized to be required.44 Administrating pamidronate or zoledronate after allo-HSCT appears to be a sensible strategy to further improve γδ T-cell-mediated anti-EBV control. All but 1 EBV reactivations were detected before the development of posttransplant lymphoproliferative disorder and treated preemptively with rituximab. Future regimens might benefit from adding CD19 to the depletion protocol. This has been done before within the context of haploidentical transplantations.14,16,19
The timing and dosing of the conditioning agents has great impact on early IR. For that reason, we individualized many parameters within the αβ T-cell depletion transplantation protocol, such as the number of αβT cells, dose of busulfan,22,51 and very early ATG administration, to avoid the presence of ATG at the time of transplantation.20,21 However, early IR was still very heterogeneous. Fludarabine is a parameter that has not yet been fully controlled in our study. We have recently shown that fludarabine exposure after allo-HSCT is highly variable, and that high fludarabine levels after allo-HSCT associate with a decreased IR, higher incidence of CMV, and impaired OS.52,53 To test whether individualized fludarabine dosing can further reduce infectious complications within the first 100 days, we started a randomized multicenter trial (NTR7136). To improve tumor control and allow for even earlier DLI, a more defined T-cell product, like a CD45RA-depleted DLI, has been shown to be safe after haploidentical HSCT, but the impact on clinical outcomes still needs to established.54
In summary, we demonstrate that a combination of early ATG, a myeloablative-reduced toxicity conditioning regimen with fludarabine, dose-adapted busulfan, and an αβ T-cell–depleted allograft of PBSCs derived from related and unrelated donors (including 9/10), is associated with a favorable risk-benefit ratio in an adult cohort, with a median age of 59 years. This platform provides a further window of opportunity for additional and early immune interventions, if needed, to further control viral toxicities in particular, as well as to further improve the control of the underlying hematological malignancy.
For original data, please contact the corresponding author (j.h.e.kuball@umcutrecht.nl).
Acknowledgments
Funding for this study was provided by the Dutch Cancer Society (KWF; UU 2015-7553) (M.A.d.W.) and Netherlands Organisation for Health Research and Development (ZonMW; 43400003), Experienced Researchers (VIDI)-ZonMW 917.11.337, KWF UU 2010-4669, UU 2013-6426, UU 2014-6790, UU 2015-7601, UU 2018-11393, UU 2018-11979, UU 2020-12586, UU 2021-13043, Vrienden van het UMCU (Friends UMC Utrecht), Association for International Cancer Research (AICR; grants 10-0736, and 15-0049) (J.K.).
Authorship
Contribution: M.A.d.W., R.A., J.-.J.B., and J.K. designed the study; M.A.d.W., A.J., and J.K. analyzed the data; F.K., L.S., and T.S. performed NK cell analysis; M.A.d.W., A.v.R., L.v.d.W., M.C.M., E.P., R.A.P.R., C.J.M.H., and J.K. included and treated patients in the study; K.N. managed the study; K.W. was responsible for graft engineering; and M.A.d.W., A.J., C.J.M.H., and J.K. wrote the manuscript.
Conflict-of-interest disclosure: J.-J.B. reports honoraria received from Avrobio, Magenta, Advanced Clinical, Takeda, and Bluerock for consulting. J.K. reports grants from Gadeta, Novartis, and Miltenyi Biotech and is inventor on patents dealing with γδ T-cell–related aspects as well as cofounder and shareholder of Gadeta. The remaining authors declare no competing financial interests.
Correspondence: Jürgen Kuball, University Medical Centre Utrecht, PO Box 85500, 3508 GA Utrecht, The Netherlands; e-mail: j.h.e.kuball@umcutrecht.nl.

