

Key Points
t(11;22)(q13;q11)/CCND1-IGL resulted in truncation of the CCND1 3′ coding sequences in a case of blastoid variant mantle cell lymphoma.
The translocation generated CCND1 with a C-terminal alteration, leading to negative CCND1 immunohistochemistry by SP4 monoclonal antibody.
Introduction
Mantle cell lymphoma (MCL) is a mature B-cell neoplasm usually composed of monomorphic small to medium-sized lymphoid cells.1 However, lymphoma cells can exhibit a variable appearance among cases, ranging from the classical morphology to lymphoblast-like cells (blastoid variant) and even occasionally to pleomorphic cells in a single case.1,2 Nevertheless, despite the variable histopathology, as t(11;14)(q13;q32) that leads to juxtaposition of the cyclin D1 (CCND1) gene with the immunoglobulin heavy (IGH) locus and overexpression of CCND1 is observed in >95% of MCL cases,1 detection of the translocation by cytogenetic methods and CCND1 expression by immunohistochemistry (IHC) readily leads to the diagnosis of MCL.
Variant translocations between CCND1 and immunoglobulin κ or λ locus (IGL) have rarely been described.3-6 In contrast to t(11;14)(q13;q32), breakage of t(11;22)(q13;q11)/CCND1-IGL was shown to occur within the 3′ untranslated region (UTR) of CCND1 exon 5, followed by IGL constant gene (IGLC) in the same transcriptional orientation.7 On the other hand, point mutations, deletions, or translocations that affect the 3′ UTR of CCND1 result in the expression of truncated messenger RNA (mRNA), which is more stable than the normal transcripts, thereby increasing the level of CCND1.8-11 Very recently, an unbalanced CCND1-IGH translocation was identified by whole-genome sequencing in which virtually the entire 14q arm is inserted at 3′ UTR of CCND1, placing the IGH enhancer near CCND1.12 In this report, we describe a case of blastoid variant MCL associated with t(11;22)(q13;q11) that affected the expression of CCND1 recognized by IHC.
Case description
A man in his 60s presented with a rapidly growing tumor in his right submandibular region. 18F-fluorodeoxyglucose positron emission tomography demonstrated marked accumulation of the tracer in the tumor; no other sites of involvement were detected. Blood cell counts, the white cell differential, and blood chemistry results were normal. The bone marrow comprised normal hematopoietic precursors.
A biopsy specimen of the tumor showed the diffuse proliferation of medium-sized lymphoid cells associated with starry-sky macrophages (Figure 1A-B). Lymphoma cells showed lymphoblastoid features characterized by nuclei with an irregular contour, dispersed chromatin, and distinct, but not prominent, nucleoli. The cells were positive for CD20, CD5, SOX11, BCL2, and MUM1 and negative for CD10 and BCL6. Over 40% of lymphoma cell nuclei were stained positive for MYC. The Ki-67 index was estimated to be 80%, and p53+ cells were below the cut-off predicting TP53 mutation by IHC (Figure 1C-F; supplemental Figure 1). CCND1 IHC using the rabbit monoclonal antibody, clone SP4,13,14 was negative, while another CCND1 IHC using the mouse monoclonal antibody clone P2D11F1114 showed strong positivity of lymphoma cell nuclei (Figure 1G-H). Flow cytometry revealed cell-surface expression of μ, δ, and κ immunoglobulins and confirmed CD5+, CD10−, and CD20+. CD23 was negative (supplemental Figure 2). We obtained the entire length of an IGH V-D-J sequence in frame and found a total of 3 mismatches compared with the closest germline sequences; the overall sequence identity was 99% (supplemental Figure 3), matching the unmutated IGH variable gene (IGHV) criteria.15 In summary, the patient had stage I MCL with histopathological features of the blastoid variant that developed and progressed through the classic MCL pathway, 1,2,16 in contrast to a previously reported patient with t(11;22) who presented with leukemic nonnodal MCL.5
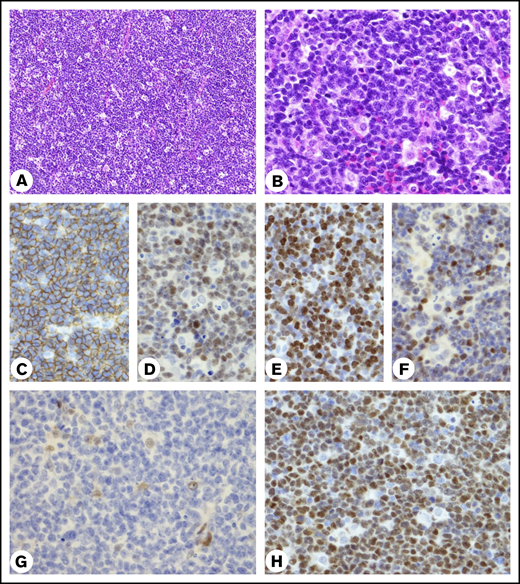
Histopathology of the biopsy specimen. (A-B) Hematoxylin and eosin–stained biopsy specimen. (A) Small vessels are seen. (B) Infiltrating tingible-body macrophages contain only a few apoptotic bodies in the cytoplasm. Original magnification of objective lens ×10 (A) and ×40 (B). (C-H) IHC results for CD20 (C), SOX11 (D), Ki-67 (E), MYC (F), CCND1 (G) by rabbit SP4 (Nichirei Biosciences, Tokyo, Japan) and CCND1 (H) by mouse P2D11F11 (Leica Biosystems, Newcastle Upon Tyne, United Kingdom). Original magnification ×40. (G) Nuclei of histiocytes and endothelial cells were stained positive for the antibody.
Histopathology of the biopsy specimen. (A-B) Hematoxylin and eosin–stained biopsy specimen. (A) Small vessels are seen. (B) Infiltrating tingible-body macrophages contain only a few apoptotic bodies in the cytoplasm. Original magnification of objective lens ×10 (A) and ×40 (B). (C-H) IHC results for CD20 (C), SOX11 (D), Ki-67 (E), MYC (F), CCND1 (G) by rabbit SP4 (Nichirei Biosciences, Tokyo, Japan) and CCND1 (H) by mouse P2D11F11 (Leica Biosystems, Newcastle Upon Tyne, United Kingdom). Original magnification ×40. (G) Nuclei of histiocytes and endothelial cells were stained positive for the antibody.
We treated the patient with 4 cycles of cyclophosphamide, doxorubicin, vincristine, and prednisolone in combination with rituximab, followed by consolidated radiotherapy. The patient has been free from relapse for 1 year after presentation.
Methods
Cytogenetic studies were performed as described previously.17 The probes for fluorescence in situ hybridization (FISH) were a Vysis LSI IGH/CCND1 XT dual-fusion (DF) translocation probe; CCND1, IGH, MYC, BCL2, and BCL6 dual-color break-apart (BA) probes; a TP53/CEP 17 FISH probe (Abbott Laboratories Abbott Park, IL), and an XL 22q11 IGL BA probe (MetaSystems, Altlussheim, Germany).17 Long-distance polymerase chain reaction (PCR) amplifying the breakpoint junctions of translocations in B-cell tumors was as described previously.17,18 PCR procedures were carried out in a Veriti 96-well thermal cycler (Applied Biosystems, Forester City, CA), and PCR products were sequenced using an ABI 310 automated sequencer (Applied Biosystems).17 This study was performed in accordance with the ethical standards presented by the institutional review board in Tenri Hospital.
Results and discussion
G-banding demonstrated the variant t(11;22)(q13;q11), instead of the standard t(11;14)(q13;q32), resulting in the characteristic 11q− and 22q+ morphology (Figure 2A). Additional abnormalities included add(6), −9, and 2 unknown marker chromosomes. Chromosomal loci for CCND2 (12p13) and CCND3 (6p21) were not affected.
![Cytogenetic analysis and molecular anatomy of t(11;22)(q13;q11)/CCND1-IGL. (A) G-banding karyotype showing t(11;22)(q13;q11) (arrows). The karyotype was 47,XY,add(6)(q21),−9,t(11;22)(q13;q11),+2mar[12]/46,XY[8]. (B) FISH of chromosome preparations using the IGH/CCND1 DF probe, consisting of red-labeled CCND1 and green-labeled IGH. G-banding and FISH picture captured through the triple-bandpass filter are aligned. Hybridization signals are indicated by arrowheads of their respective colors. (C) FISH using the CCND1 BA probe, consisting of green-labeled centromeric 5′ CCND1 and red-labeled telomeric 3′ CCND1, followed by the IGL BA probe, consisting of red-labeled centromeric 5′ IGL and green-labeled telomeric 3′ IGL. G-banding and FISH captured through the rhodamine and FITC filters are aligned, and hybridization signals are indicated by arrowheads of their respective colors. (D) Top: Schematic diagram of 11q13/CCND1 and 22q11/IGL in the centromere (cen) to telomere (tel) orientation. Sequences of the primers for CCND1 (forward) and IGL (reverse) are described in supplemental Figure 5. Breakpoints are indicated by open arrows. The gray open arrows are the positions of the breakpoints determined by Komatsu et al (chr11:69 653,420/1 and chr22:22 905,055/6).7 Bottom: Nucleotide sequences of the CCND1-IGL junction (accession number LC582849) and deduced amino acids designated by the single-letter code. Twenty-two nontemplated nucleotides inserted at the junction are underlined. Vertical lines indicate nucleotide identity. Each breakpoint was chr11:69 651,255/6 and chr22:22 904,994/5 (open arrows). Amino acid residues reported to be mutated in MCL or non-hematologic cancers are underlined.19 FITC, fluorescein isothiocyanate.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/1/10.1182_bloodadvances.2020003417/2/m_advancesadv2020003417f2.png?Expires=1767720526&Signature=BKYeSlraKtmGasjJH31GMFz4PazBmTBgeaju4E5ji8XhxFTH1uUk1-1a-1hCyDt5siR6NXfILY1m~ji7oLF1QPBc~Sqq1S9naNJlGuSCnkr~4FzSk01O-GzTQLkR7Q6VPzuz-FHtZg78n0ecy-XV5gKkrRWRZFCce3aLircxfiFwoegTwe~SCC55NaOEt~MyJhO8KUUvEoJpKeKKSVkG~2JEJmIKhBlj8A~kwzp9mPRwE16s-9jQAaSuRVN3IdNjhAnaibGuK~0ujZ03~2sm6yJguAZJdV78nJFUwYUdmJhmWMaHaf87PtFMtJhYB41uLq-rUUtnVBu05IYMeFS4Dw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Cytogenetic analysis and molecular anatomy of t(11;22)(q13;q11)/CCND1-IGL. (A) G-banding karyotype showing t(11;22)(q13;q11) (arrows). The karyotype was 47,XY,add(6)(q21),−9,t(11;22)(q13;q11),+2mar[12]/46,XY[8]. (B) FISH of chromosome preparations using the IGH/CCND1 DF probe, consisting of red-labeled CCND1 and green-labeled IGH. G-banding and FISH picture captured through the triple-bandpass filter are aligned. Hybridization signals are indicated by arrowheads of their respective colors. (C) FISH using the CCND1 BA probe, consisting of green-labeled centromeric 5′ CCND1 and red-labeled telomeric 3′ CCND1, followed by the IGL BA probe, consisting of red-labeled centromeric 5′ IGL and green-labeled telomeric 3′ IGL. G-banding and FISH captured through the rhodamine and FITC filters are aligned, and hybridization signals are indicated by arrowheads of their respective colors. (D) Top: Schematic diagram of 11q13/CCND1 and 22q11/IGL in the centromere (cen) to telomere (tel) orientation. Sequences of the primers for CCND1 (forward) and IGL (reverse) are described in supplemental Figure 5. Breakpoints are indicated by open arrows. The gray open arrows are the positions of the breakpoints determined by Komatsu et al (chr11:69 653,420/1 and chr22:22 905,055/6).7 Bottom: Nucleotide sequences of the CCND1-IGL junction (accession number LC582849) and deduced amino acids designated by the single-letter code. Twenty-two nontemplated nucleotides inserted at the junction are underlined. Vertical lines indicate nucleotide identity. Each breakpoint was chr11:69 651,255/6 and chr22:22 904,994/5 (open arrows). Amino acid residues reported to be mutated in MCL or non-hematologic cancers are underlined.19 FITC, fluorescein isothiocyanate.
Cytogenetic analysis and molecular anatomy of t(11;22)(q13;q11)/CCND1-IGL. (A) G-banding karyotype showing t(11;22)(q13;q11) (arrows). The karyotype was 47,XY,add(6)(q21),−9,t(11;22)(q13;q11),+2mar[12]/46,XY[8]. (B) FISH of chromosome preparations using the IGH/CCND1 DF probe, consisting of red-labeled CCND1 and green-labeled IGH. G-banding and FISH picture captured through the triple-bandpass filter are aligned. Hybridization signals are indicated by arrowheads of their respective colors. (C) FISH using the CCND1 BA probe, consisting of green-labeled centromeric 5′ CCND1 and red-labeled telomeric 3′ CCND1, followed by the IGL BA probe, consisting of red-labeled centromeric 5′ IGL and green-labeled telomeric 3′ IGL. G-banding and FISH captured through the rhodamine and FITC filters are aligned, and hybridization signals are indicated by arrowheads of their respective colors. (D) Top: Schematic diagram of 11q13/CCND1 and 22q11/IGL in the centromere (cen) to telomere (tel) orientation. Sequences of the primers for CCND1 (forward) and IGL (reverse) are described in supplemental Figure 5. Breakpoints are indicated by open arrows. The gray open arrows are the positions of the breakpoints determined by Komatsu et al (chr11:69 653,420/1 and chr22:22 905,055/6).7 Bottom: Nucleotide sequences of the CCND1-IGL junction (accession number LC582849) and deduced amino acids designated by the single-letter code. Twenty-two nontemplated nucleotides inserted at the junction are underlined. Vertical lines indicate nucleotide identity. Each breakpoint was chr11:69 651,255/6 and chr22:22 904,994/5 (open arrows). Amino acid residues reported to be mutated in MCL or non-hematologic cancers are underlined.19 FITC, fluorescein isothiocyanate.
To confirm that t(11;22) involved CCND1, we hybridized the metaphase spreads with the IGH/CCND1 DF probe, showing that der(22)t(11;22) at q11 was marked by the red-labeled CCND1 probe, while no CCND1-IGH fusion signals were generated. As the red signal on der(22)t(11;22) was small, breakage was considered to occur at a point close to the telomeric end of the CCND1 probe. Accordingly, interphase nuclei carried 2 red, 1 small red, and 2 green hybridization signals (Figure 2B). Next, we hybridized the metaphase spreads serially with the CCND1 BA and IGL BA probes. As shown in Figure 2C, der(11)t(11;22) was marked by the green-labeled centromeric CCND1 and telomeric IGL probes, and der(22)t(11;22) was marked by the red-labeled centromeric IGL and telomeric CCND1 probes, respectively, confirming the generation of the fusion gene between 5′ CCND1 and 3′ IGL sequences at q13 of der(11)t(11;22) and its reciprocal between 5′ IGL and 3′ CCND1 sequences at q11 of der(22)t(11;22), both in the centromere to telomere orientation. No BA signals or increased copies of the IGH, MYC, BCL2, or BCL6 gene were found. No 17p/TP53 deletion was detected by FISH (supplemental Figure 4).
To amplify the CCND1-IGL junction, we employed long-distance PCR using primers complementary to CCND1 exon 5 and to the intronic sequences immediately 5′ of exon 5 and those to the consensus sequences of IGLC, generating ∼2-kb PCR products (supplemental Figure 5). Nucleotide sequencing of the products revealed that the breakpoint on the CCND1 side was 24 bp 5′ of the stop codon of the coding sequence, and that on the IGL side was 7 bp 3′ of the 5′ end of the IGLJ3 segment, and 22 nontemplated nucleotides were inserted at the junction (Figure 2D), indicating that the t(11;22)/CCND1-IGL developed through the V-(D)-J recombination mechanism in the precursor B-cell stage, similar to t(11;14)/CCND1-IGH in the majority of MCL cases.12 As a result of translocation, the 8 C-terminal amino acids of CCND1 were theoretically replaced by 4 unrelated amino acid residues.
Chromosomal translocations between oncogenes and immunoglobulin genes in mature B-cell tumors, in principle, do not disrupt the protein coding regions of the relevant oncogenes, producing intact oncogene products. In contrast, we found that t(11;22) disrupted the CCND1 coding sequences, affecting the C terminus of the protein product, leading to the absence of the immunoreactivity against SP4, which was developed by immunization with synthetic polypeptide from the C terminus of human CCND1,19 but retaining the P2D11F11 reactivity directing full-length CCND1.20 Alternatively, the translocation may have promoted restrictive transcription of the short-form CCND1 mRNA, under the influence of the translocated IGL enhancer activity, and generated abundant CCND1 protein composed of 274 amino acids that differs at the C terminus from the complete 294 amino acid protein,11 thus lacking the epitope for SP4 recognition; this possibility is consistent with the fact that short-form mRNA is associated with the blastoid variant histopathology and a high Ki-67 index.21-23
Negative SP4 immunostaining despite the presence of t(11;14) was previously described in 2 patients19 ; one carried a mutation (D292P) in the C-terminal region of CCND1 (Figure 2D), which was considered to impair SP4 binding, and the other lacked long-form CCND1 mRNA but exclusively expressed short-form CCND1 mRNA by an unknown mechanism. Taken together with the results of these 2 cases and our current case, the combination of CCND1 IHC using SP4 and P2D11F11 and FISH testing using DF and BA probes is the best approach for MCL diagnosis to avoid missing any of the possible variants and false-negative results (supplemental Figure 6).
The data for the sequences representing the CCND1-IGL junction reported in this article have been deposited in the DNA Data Bank of Japan database (accession number LC582849).
Requests for data sharing should be e-mailed to the corresponding author, Hitoshi Ohno (hohno@tenriyorozu.jp).
Acknowledgment
This work was supported by the Tenri Foundation.
Authorship
Contribution: F.M. and H.O. collected data, constructed the figures, and wrote the manuscript; C.K., M.N., K. Takeoka, K.F., and M.H. performed data analysis; F.I. treated the patient; M.F., K. Takeuchi, and H.N. reviewed the histopathology; and all authors reviewed and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Hitoshi Ohno, Department of Hematology, Tenri Hospital, 200 Mishima, Tenri, Nara 632-8552, Japan; e-mail: hohno@tenriyorozu.jp.

