

Key Points
The novel ROR1-targeting antibody-drug conjugate huXBR1-402-G5-PNU effectively suppresses growth of ROR1+ tumor cells in vitro and in vivo.
HuXBR1-402-G5-PNU exhibits BCL2-dependent synergy with the BCL2-inhibitor venetoclax in ROR1+ leukemia cells.

Abstract
Antibody-drug conjugates directed against tumor-specific targets have allowed targeted delivery of highly potent chemotherapy to malignant cells while sparing normal cells. Receptor tyrosine kinase-like orphan receptor 1 (ROR1) is an oncofetal protein with limited expression on normal adult tissues and is overexpressed on the surface of malignant cells in mantle cell lymphoma, acute lymphocytic leukemia with t(1;19)(q23;p13) translocation, and chronic lymphocytic leukemia. This differential expression makes ROR1 an attractive target for antibody-drug conjugate therapy, especially in malignancies such as mantle cell lymphoma and acute lymphocytic leukemia, in which systemic chemotherapy remains the gold standard. Several preclinical and phase 1 clinical studies have established the safety and effectiveness of anti-ROR1 monoclonal antibody–based therapies. Herein we describe a humanized, first-in-class anti-ROR1 antibody-drug conjugate, huXBR1-402-G5-PNU, which links a novel anti-ROR1 antibody (huXBR1-402) to a highly potent anthracycline derivative (PNU). We found that huXBR1-402-G5-PNU is cytotoxic to proliferating ROR1+ malignant cells in vitro and suppressed leukemia proliferation and extended survival in multiple models of mice engrafted with human ROR1+ leukemia. Lastly, we show that the B-cell lymphoma 2 (BCL2)-dependent cytotoxicity of huXBR1-402-G5-PNU can be leveraged by combined treatment strategies with the BCL2 inhibitor venetoclax. Together, our data present compelling preclinical evidence for the efficacy of huXBR1-402-G5-PNU in treating ROR1+ hematologic malignancies.
Introduction
Conventional chemotherapeutic drugs lack a significant therapeutic window and are often associated with significant adverse events. Monoclonal antibodies targeting tumor-specific antigens may mitigate off-target effects of conventional chemotherapy but are dependent on the intracellular domain for signaling and the extracellular domain for mediating cellular cytotoxicity, for which resistance often develops.1,2 Novel antibody-drug conjugates (ADCs) exploit cancer-specific antigens to deliver highly potent, cytotoxic payloads to tumor cells. Clinically, 7 ADCs are approved for cancer treatment, including brentuximab vedotin for Hodgkin lymphoma in 2011 and ado-trastuzumab emtansine in 2013 for metastatic breast cancer.3,4 Currently, >175 ADCs are in multiple stages of investigation, from preclinical studies to early-phase clinical trials.5
Receptor tyrosine kinase-like orphan receptor 1 (ROR1) is a surface transmembrane receptor tyrosine kinase that is overexpressed in multiple malignancies, including B-cell acute lymphoblastic leukemia (B-ALL), mantle cell lymphoma (MCL), and chronic lymphocytic leukemia (CLL).6-9 In contrast, ROR1 is expressed at low levels on hematogones and absent on most adult tissues.6,8,10 This tumor-specific overexpression and limited expression on normal tissues have made ROR1 a popular candidate for therapeutics that can target cancer cells while sparing normal tissues.
Multiple preclinical studies have explored ROR1 as a targetable antigen. Targeting mechanisms include small molecule inhibitors, immunoliposomes, immunotoxins, bispecific antibodies, chimeric-antigen receptor modified (CAR)-T cells, ROR1 peptide vaccines, and monoclonal antibodies.10-20 Early phase 1 clinical studies of cirmtuzumab, an anti-ROR1 monoclonal antibody (UC-961 clone), in patients with CLL showed that it was safe in patients without dose-limiting toxicities.18 Although cirmtuzumab failed to eradicate disease, this early trial established ROR1 as a safe therapeutic target.
Here, we evaluate a first-in-class ROR1-targeted ADC, huXBR1-402-G5-PNU. huXBR1-402 is a humanized anti-ROR1 monoclonal antibody derived from rabbit anti-human ROR1 monoclonal antibody XBR1-402 that is conjugated to a derivative of PNU-159682, a highly potent metabolite of the parent anthracycline nemorubicin.21-23 This strategy combines the targeting ability of the anti-ROR1 antibody with the cytotoxic effect of the payload.24 We evaluated the effects of huXBR1-402-G5-PNU on classical ROR1+, highly proliferative hematologic malignancies, including B-ALL and MCL, exhibiting both in vitro cytotoxicity and in vivo disease control.
Materials and methods
ADC therapeutics
Human samples and study approval
Peripheral blood mononuclear cells (PBMCs) were obtained from normal donors or patients with CLL in accordance with the Declaration of Helsinki. All subjects have given written informed consent for the blood products to be used for research under an institutional review board–approved protocol. Blood from patients with CLL was collected at The Ohio State University Comprehensive Cancer Center (Columbus, OH). Normal cells were obtained from Red Cross partial leukocyte preparations. PBMCs were isolated via density-gradient centrifugation by using Ficoll-Paque Plus (GE Healthcare, Uppsala, Sweden). CLL cells were selected by negative selection using B-cell RosetteSep enrichment kits (#15068; Stemcell Technologies, Vancouver, BC, Canada) according to the manufacturer’s protocol.
Cell culture
For all primary cell and cell line experiments, unless otherwise stated, cells were cultured in full serum media defined as 37°C and 5% carbon dioxide (CO2) in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum, 2 mM l-glutamine (#15030164; Invitrogen, Carlsbad, CA), and 56 U/mL and 56ug/mL of penicillin and streptomycin respectively (#15140122; Invitrogen).
Surface ROR1 antigen staining and quantification
For competitive antibody binding assays, cell lines were incubated with primary antibody against ROR1 (clones, huXBR1-402, or 2A2), trastuzumab (isotype control), or in the absence of antibody (negative control). Alternatively, ROR1 was detected with directly labeled anit-ROR1 antibodies: 2A2-PE (monoclonal; #357804; BioLegend, San Diego, CA) or ROR1-PE (polyclonal; #FAB2000P; R&D Systems, Minneapolis, MN).
For antigen detection with huXBR1-402, total PBMCs from patients with CLL (n = 14) or normal donors (n = 8) were isolated and stained with anti-human CD19 BV510, anti-human CD5 PeCy7, near-IR Fixable Viability stain, and either huXBR1-402 or trastuzumab (isotype). Matched CLL T-cell staining values were reported if present in sample (at least 10 000 cells). Secondary antibody detection was done with Alexa Fluor 647 AffiniPure Goat Anti-Human IgG, Fc Fragment Specific (#NC0248893; Jackson ImmunoResearch Laboratories, West Grove, PA).
Quantification of ROR1 surface expression was performed by using the QuantiBRITE PE bead assay (#340495; BD Biosciences) and ROR1 (clone: 2A2)-PE (#357804; BioLegend).
Viability and cell cycling assays
HG3, JeKo-1, Mino, 697, Kasumi-2, REH, and MEC1 cell lines obtained from ATCC were serum starved for 16 hours and reintroduced to full serum media supplemented with 10 µg/mL huXBR1-402-G5-PNU and relevant controls for 72 hours.
Viability.
Cells were stained with Annexin V-FITC (Annexin V) and propidium iodide (#A432; Leinco Technologies, St. Louis, MO). Tumor cells from patients with CLL (n = 11) were isolated as described earlier and treated the same as cell lines. Data are reported as the percentage of viable Annexin V and propidium iodide double-negative cells normalized to vehicle control.
Cell cycle.
Cells were stained with Click-iT Plus EdU Alexa Fluor 647 Flow Cytometry Assay Kit (#C10635; Invitrogen) and FxCycle Violet Ready Flow Reagent (#R37166; Invitrogen). Data are reported as the percentage of cells in G0/G1 (5-ethynyl-2′-deoxyuridine negative [EdU–] and FxCycle Violet–), S phase (EdU+ positive), and G2/M (EdU–, FxCycle Violet+).
Flow cytometry
All flow cytometric experiments were performed by using Beckman Coulter Gallios flow cytometers (Brea, CA). Fluorochrome-labeled human monoclonal antibodies included: anti-human CD19-BV510 (HIB19), CD5-PE/Cy7 (UCHT2) (BioLegend), and LIVE/DEAD Fixable Near-IR Dead Cell Stain (Invitrogen). Fluorochrome-labeled murine antibodies included anti-mouse CD5 BV421 (52-7.3), CD19 PE (6D5), and CD45 APC (30-F11) (BioLegend). Absolute cell concentrations were obtained by quantitative flow cytometry using CountBright absolute counting beads (Invitrogen).
Mouse leukemia engraftment
Human pre–B-ALL 697 cells were harvested and resuspended in sterile phosphate-buffered saline for injection. Immunodeficient NOD-SCID mice 6 to 8 weeks old received an intravenous tail vein injection of 100 µL containing 5 × 106 live 697 cells on day 0. Due to the aggressive disease course, treatment with 1 mg/kg huXBR1-402-G5-PNU or trastuzumab-G5-PNU was given through intravenous injection on days 7 and 14 postengraftment (n = 6 mice per group). Mice were monitored for overall survival.
Splenocytes from heavily leukemic (>80% ROR1+/CD5+/CD19+ cells in spleen) hROR1 × TCL1 donor C57BL/6 mice as previously described were isolated and resuspended in sterile phosphate-buffered saline for injection.12 Healthy age-matched 6- to 8-week-old female C57BL/6J mice from The Jackson Laboratory (Bar Harbor, ME) received an intravenous lateral tail vein injection of 100 µL containing 5 × 106 live splenocytes (>85% CD5+/CD19+ leukemic cells). Mice were stratified and randomly selected to the following treatment groups after leukemia onset, defined as when >5% of CD45+ cells in the peripheral blood consisted of CD5+CD19+ leukemic B cells. Treatment with 1 mg/kg huXBR1-402-G5-PNU, huXBR1-402, or trastuzumab-G5-PNU was given through intraperitoneal injections on days 1, 3, and 5 postrandomization (n = 9 mice per group). Mice were monitored for overall survival and disease progression by flow cytometry on a weekly basis.
Humane animal treatment practices were followed. Mice were killed with CO2 upon reaching Institutional Animal Care and Use Committee criteria for early removal from the study (eg, labored breathing, weight loss >20% over 1 week, severe lethargy). All animal experiments were completed under an approved IACUC protocol.
Loewe additivity assays
Using a 96-well plate, 20 000 HG3, 697 (neo and B-cell lymphoma 2 [BCL2]), Mino, or JeKo-1 cells were treated with varying doses of venetoclax in combination with either huXBR1-402-G5-PNU or trastuzumab-G5-PNU (isotype control) for 72 hours. Cells were incubated with CellTiter 96 AQueous MTS reagent (#G1111; Promega, Madison, WI) at 37°C, 5% CO2 for 3 to 6 hours after drug incubation. Spectral absorbance at 490 nm was measured by using a DTX 880 plate reader.
Immunoblots
A total of 25 µg of cell lysates was loaded onto 4% to 20% Mini-PROTEAN TGX Precast Protein Gels (Bio-Rad, Hercules, CA; catalog #4561094) and run at 100 V for 2 hours. Gels were transferred onto nitrocellulose membranes by using the Bio-Rad Trans-Blot Turbo Transfer System (catalog #1704150). Primary BCL2 antibody (Agilent; catalog #M0887) was used at a dilution of 1:1000 in BLOTTO (Thermo Fisher Scientific, Waltham, MA; catalog #37530) and probed with anti-mouse immunoglobulin G, horseradish peroxidase (HRP)-linked secondary antibody (Cell Signaling Technologies; catalog #7076). Blots were stripped with Restore PLUS Western Blot Stripping Buffer (Thermo Fisher Scientific) for 10 minutes before probing with β-actin (housekeeping control) primary antibody (Cell Signaling Technologies; catalog #3700) at a concentration of 1:1000 in BLOTTO. Secondary staining was performed with anti-mouse immunoglobulin G, HRP-linked secondary antibody (Cell Signaling Technologies; catalog #7076). Detection of HRP was performed with Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific; catalog #32106), and blots were exposed for 30 to 60 seconds.
Statistical analysis
R statistical software (R Foundation for Statistical Computing, Vienna, Austria) and GraphPad Prism 8 software (GraphPad Software, La Jolla, CA) were used to aggregate and analyze all data. Kaluza Analysis Software by Beckman Coulter was used to analyze all flow cytometry data (Brea, CA). Student t tests were used to analyze all panels of Figures 1 and 6A. For multi-group comparisons (including murine studies), mixed effect models were used for data analysis, incorporating repeated measures for each subject, with group (treatment groups) and time as factors to account for dependencies among different groups. Tukey’s post hoc analysis was used to obtain P values for multiple comparisons. Significant P values are set to <0.05. For in vivo survival studies, log-rank tests were used to compare survival probabilities across different treatment groups. Loewe additivity and effective dose concentrations were modeled by using Combenefit.26
Results
Humanized huXBR1-402 binds human ROR1
The huXBR1-402 antibody is a humanized version of human monoclonal ROR1 monoclonal antibody XBR1-402 generated through phage display libraries and validated with next-generation sequencing. From previous studies, we determined that XBR1-402 bound to the immunoglobulin and frizzled domain of ROR1.23 We therefore first sought to evaluate the binding specificity of the humanized huXBR1-402 antibody by assessing the ability of the huXBR1-402 ROR1 antibody clone to block the binding of commercial ROR1 antibodies with known epitopes. We first incubated ROR1+ MCL lines JeKo-1 and Mino cells with unconjugated monoclonal huXBR1-402, unconjugated mouse monoclonal ROR1 antibody (clone, 2A2), trastuzumab (anti-HER2 isotype control), or left the cells unstained. We showed that blocking with huXBR1-402 but not trastuzumab inhibited subsequent binding of phycoerythrin (PE) conjugated 2A2 monoclonal ROR1 antibody (2A2-PE) on the surface of JeKo-1 and Mino cells (Figure 1A). Blocking with huXBR1-402 or 2A2 ROR1 antibodies did not inhibit binding of the PE-conjugated goat polyclonal ROR1 antibody (ROR1-PE), likely because polyclonal antibodies bind to multiple ROR1 epitopes. Similarly, staining with unconjugated ROR1 polyclonal antibody can inhibit the subsequent binding of the 2A2 monoclonal ROR1 antibody (supplemental Figure 1). Because ROR1 is highly expressed in 95% of CLL cases, we assessed binding of the huXBR1-402 in primary CLL patient samples. Compared with matched patient T cells, a twofold increase of huXBR1-402 binding to leukemic cells was observed, and this was confirmed by surface quantification of ROR1 (Figure 1B-C). We also found that ROR1 is overexpressed on malignant CLL cells compared with B cells from healthy donors (Figure 1D).

Novel anti-ROR1 antibody clone huXBR1-402 binds specifically to human ROR1 overexpressed on leukemic B cells. (A) Antibody competitive binding assay showing results from ROR1+ Mino, JeKo-1 cells, and ROR1– MEC1 cells. Cells were preincubated with either huXBR1-402, 2A2, trastuzumab (negative), or a no-block control indicated immediately below the x-axis. Cells are then probed with fluorescence-conjugated ROR1 antibodies (2A2-PE, monoclonal ROR1 antibody; ROR1-PE, polyclonal ROR1 antibody) indicated below the x-axis legends. n = 3 independent experiments. (B) huXBR1-402 primary antibody-stained CLL patient–derived PBMCs detected with AF647 conjugated secondary antibody against human Fc region. Staining is shown for CD19+/CD5+ CLL B cells and matched CD3+ T cells. n = 14 patients with CLL, n = 8 matched T-cell samples. (C) Quantification of surface ROR1 expression in primary CLL PBMCs. Quantification is shown for CLL B cells and matched T cells. n = 14 patients with CLL, n = 8 matched T-cell samples. (D) ROR1 staining expression in B cells from CLL and healthy donor PBMCs normalized to matched T cells. n = 8 patients with CLL, n = 7 normal donors. Data were analyzed by flow cytometry, and statistical significance was analyzed via Student t tests. MFI, mean fluorescence intensity. Error bars indicate ± SE.
Novel anti-ROR1 antibody clone huXBR1-402 binds specifically to human ROR1 overexpressed on leukemic B cells. (A) Antibody competitive binding assay showing results from ROR1+ Mino, JeKo-1 cells, and ROR1– MEC1 cells. Cells were preincubated with either huXBR1-402, 2A2, trastuzumab (negative), or a no-block control indicated immediately below the x-axis. Cells are then probed with fluorescence-conjugated ROR1 antibodies (2A2-PE, monoclonal ROR1 antibody; ROR1-PE, polyclonal ROR1 antibody) indicated below the x-axis legends. n = 3 independent experiments. (B) huXBR1-402 primary antibody-stained CLL patient–derived PBMCs detected with AF647 conjugated secondary antibody against human Fc region. Staining is shown for CD19+/CD5+ CLL B cells and matched CD3+ T cells. n = 14 patients with CLL, n = 8 matched T-cell samples. (C) Quantification of surface ROR1 expression in primary CLL PBMCs. Quantification is shown for CLL B cells and matched T cells. n = 14 patients with CLL, n = 8 matched T-cell samples. (D) ROR1 staining expression in B cells from CLL and healthy donor PBMCs normalized to matched T cells. n = 8 patients with CLL, n = 7 normal donors. Data were analyzed by flow cytometry, and statistical significance was analyzed via Student t tests. MFI, mean fluorescence intensity. Error bars indicate ± SE.
huXBR1-402-G5-PNU induces apoptosis in actively proliferating cells
PNU-159682 is an anthracycline derivative that, in addition to DNA intercalation, inhibits topoisomerase I, which is essential for DNA replication.27,28 We therefore hypothesized that huXBR1-402-G5-PNU targets proliferating cells. We tested the effects of huXBR1-402-G5-PNU in aggressive, proliferative, ROR1+ tumor cells: pre–B-ALL (697, Kasumi-2), MCL (JeKo-1 and Mino), and CLL (HG3) cell lines (supplemental Figure 2). ROR1+ B-ALL cell lines Kasumi-2 and 697 showed increased sensitivity to huXBR-402-G5-PNU compared with the ROR1– ALL cell line REH (Figure 2A; supplemental Figure 3). In MCL cell lines, the viability of ROR1+JeKo-1 and Mino cells decreased significantly after treatment with huXBR1-402-G5-PNU compared with trastuzumab-G5-PNU and ROR1– MEC1 at 24, 72, and 96 hours (Figure 2B; supplemental Figure 4). In addition, huXBR1-402-G5-PNU did not exhibit specific cytotoxicity toward ROR1– and acute myeloid leukemia cell lines (supplemental Figure 5). Although CLL HG3 cell lines did not exhibit changes in viability, significant antiproliferative effects were appreciable when evaluating absolute cell counts (Figure 2C). Furthermore, cell cycle analysis via concurrent EdU incorporation and DNA staining revealed a G2/M block 72 hours’ posttreatment with huXBR1-402-G5-PNU in all ROR1+ cells but not ROR1– MEC1 cells, reminiscent of the induction of a G2/M cell cycle block reported for nemorubicin (Figure 2D).29
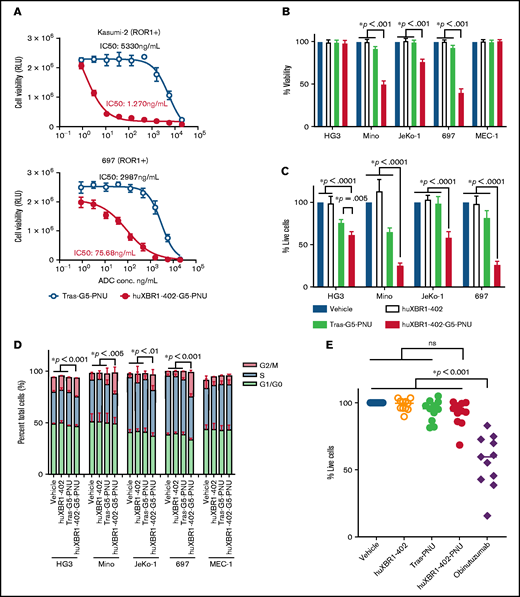
huXBR1-402-G5-PNU is cytotoxic to proliferating ROR1+ cell lines in vitro. (A) Dose response of ROR1+ pre–B-ALL cell lines Kasumi-2 and 697 to huXBR1-402-G5-PNU (red line) and trastuzumab-G5-PNU (blue line; Tras-G5-PNU). Half maximal inhibitory concentrations (IC50) + 95% confidence interval are as follows: Kasumi-2: huXBR1-402-G5-PNU, 1.270 ng/mL (0.31-5.22); Tras-G5-PNU, 5330 ng/mL (2698-10527); 697: huXBR1-402-G5-PNU, 75.68 ng/mL (50.43-113.5); Tras-G5-PNU, 2987 ng/mL (2463-3622). Viability was measured by using MTS assays, and relative light unit (RLU) values for each dose were generated after 72 hours of treatment in 2 independent experiments. (B) Direct cytotoxicity assay on ROR1+ CLL cell line HG3, MCL cell lines JeKo-1 and Mino, ALL cell line 697, and ROR1– CLL cell line MEC1. 2.5 × 105 plated cells were treated with 10 µg/mL of relevant antibodies and controls. Normalized viability (to vehicle) is reported as measured by Annexin V/propidium iodide staining after 72 hours of culture. n = at least 3 independent experiments. (C) Matched absolute cell count (normalized to vehicle) per microliter of culture for all conditions shown. n = at least 3 independent experiments. (D) Cell cycle analysis as measured by 2-hour AF647 conjugated EdU (AF647) incorporation and FxCycle Violet DNA (BV421) stain in paired samples seen in panel B. G1/G0 (green), AF647–/BV421–; S (blue), AF647+; G2/M (red), AF647-–/BV421+. n = at least 3 independent experiments. For panels C and D: red, huXBR1-402-G5-PNU; green, Tras-G5-PNU; white, unconjugated huXBR1-402; blue, vehicle treatments. (E) Direct cytotoxicity assay on primary CLL purified B cells. 1 × 106 plated cells were treated with 10 µg/mL of huXBR1-402-G5-PNU and relevant antibody isotypes (Tras-G5-PNU; unconjugated huXBR1-402) and controls (vehicle; positive control obinutuzumab). Viability (% live cells) normalized to vehicle treatment is reported as measured by Annexin V/propidium iodide staining after 72 hours of culture. n = 11 patients. All data were analyzed by flow cytometry. Statistical significance for multiple comparisons was analyzed via one-way analysis of variance with Tukey’s post hoc test. Error bars indicate ± SE.
huXBR1-402-G5-PNU is cytotoxic to proliferating ROR1+ cell lines in vitro. (A) Dose response of ROR1+ pre–B-ALL cell lines Kasumi-2 and 697 to huXBR1-402-G5-PNU (red line) and trastuzumab-G5-PNU (blue line; Tras-G5-PNU). Half maximal inhibitory concentrations (IC50) + 95% confidence interval are as follows: Kasumi-2: huXBR1-402-G5-PNU, 1.270 ng/mL (0.31-5.22); Tras-G5-PNU, 5330 ng/mL (2698-10527); 697: huXBR1-402-G5-PNU, 75.68 ng/mL (50.43-113.5); Tras-G5-PNU, 2987 ng/mL (2463-3622). Viability was measured by using MTS assays, and relative light unit (RLU) values for each dose were generated after 72 hours of treatment in 2 independent experiments. (B) Direct cytotoxicity assay on ROR1+ CLL cell line HG3, MCL cell lines JeKo-1 and Mino, ALL cell line 697, and ROR1– CLL cell line MEC1. 2.5 × 105 plated cells were treated with 10 µg/mL of relevant antibodies and controls. Normalized viability (to vehicle) is reported as measured by Annexin V/propidium iodide staining after 72 hours of culture. n = at least 3 independent experiments. (C) Matched absolute cell count (normalized to vehicle) per microliter of culture for all conditions shown. n = at least 3 independent experiments. (D) Cell cycle analysis as measured by 2-hour AF647 conjugated EdU (AF647) incorporation and FxCycle Violet DNA (BV421) stain in paired samples seen in panel B. G1/G0 (green), AF647–/BV421–; S (blue), AF647+; G2/M (red), AF647-–/BV421+. n = at least 3 independent experiments. For panels C and D: red, huXBR1-402-G5-PNU; green, Tras-G5-PNU; white, unconjugated huXBR1-402; blue, vehicle treatments. (E) Direct cytotoxicity assay on primary CLL purified B cells. 1 × 106 plated cells were treated with 10 µg/mL of huXBR1-402-G5-PNU and relevant antibody isotypes (Tras-G5-PNU; unconjugated huXBR1-402) and controls (vehicle; positive control obinutuzumab). Viability (% live cells) normalized to vehicle treatment is reported as measured by Annexin V/propidium iodide staining after 72 hours of culture. n = 11 patients. All data were analyzed by flow cytometry. Statistical significance for multiple comparisons was analyzed via one-way analysis of variance with Tukey’s post hoc test. Error bars indicate ± SE.
huXBR1-402-G5-PNU targets proliferating leukemia cells in vivo and increases survival
To evaluate the efficacy of huXBR1-402-G5-PNU on long-term leukemia control and proliferation in a complex microenvironment, we tested huXBR1-402-G5-PNU in vivo in 2 murine models of aggressive human ROR1+leukemia.
We initially used a cell line–derived xenograft model by engrafting ROR1+ human pre–B ALL cell line 697 into NOD-SCID mice to establish an aggressive disseminated disease. Two doses of XBR1-402-G5-PNU (1 mg/kg) led to a significantly prolonged survival in the XBR1-402-G5-PNU treatment group compared with the isotype control trastuzumab-G5-PNU (Figure 3A). Mice treated with XBR1-402-G5-PNU showed no gross signs of treatment-related toxicity.
![huXBR1-402-PNU suppressed leukemic growth and increased overall survival in models of aggressive human and murine ROR1+ leukemia. (A) 697 ROR1+ ALL disseminated leukemia model. Kaplan-Meier survival curve showing death of mice in each treatment group (trastuzumab-G5-PNU [Tras-G5-PNU] and XBR1-402-G5-PNU). Mice were treated intravenously with 1 mg/kg of drug on days 7 and 14 after implantation. Survival data were analyzed with log-rank test. n = 6 mice per group. (B) Schematic showing the experimental design for establishing human ROR1-expressing murine TCL1 model of CLL and intraperitoneal dosing scheme for fully humanized huXBR1-402. Briefly, C57BL/6 mice were engrafted with 5 × 106 huROR1-TCL1 spleen-derived leukemic cells via tail vein injection. All mice were treated with 1 mg/kg of either huXBR1-402-G5-PNU, Tras-G5-PNU, or equal volumes of vehicle 3 times per week for 1 week after enrollment criteria were met (peripheral leukemia proportion was >5% of total white blood cells). (C) Weekly peripheral leukemia cell counts of a single mouse cohort as determined by flow cytometry. Mixed effects model was used to analyze differences with P value given at week 6. n = 4 mice per group. (D) Kaplan-Meier survival curve showing death of mice in each treatment group (vehicle, trastuzumab-PNU, unconjugated huXBR1-402, and huXBR1-402 PNU). Survival data were analyzed by using the log-rank test. n = 9 mice per group. Error bars indicate ± SE.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/16/10.1182_bloodadvances.2020003276/5/m_advancesadv2020003276f3.png?Expires=1769432947&Signature=hRo44MA6JghEl4CBkzU3x~sFNxVKTOf8xlJWmkeavit2hb~QH8UN~Dlu3o476yHymXj510GILumSMIml7SUmsgFtHCrL2QtSiSyPEUsA6j2~aSUibWnQJXig-eT6pN2TrC1YrN7KMUTXxblXBdF22U5s95X1jIOfEVaK-x3AkbLk6C6NmIP31J7PltbrKlh4VFdS4YGwZvZQ13zhTN81i4XYfNaHQv5xPP2sVgKjSlnVMNiQQ-SSpQifZh7ZFPac1nGhqe-1UMWmyb5SCmkSvkvvEntuGu-sS-OW9HCsXN-9SSSt7EAIRqkjuXDchP7PYORf8q-rWmz0DTA45zPwKg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
huXBR1-402-PNU suppressed leukemic growth and increased overall survival in models of aggressive human and murine ROR1+ leukemia. (A) 697 ROR1+ ALL disseminated leukemia model. Kaplan-Meier survival curve showing death of mice in each treatment group (trastuzumab-G5-PNU [Tras-G5-PNU] and XBR1-402-G5-PNU). Mice were treated intravenously with 1 mg/kg of drug on days 7 and 14 after implantation. Survival data were analyzed with log-rank test. n = 6 mice per group. (B) Schematic showing the experimental design for establishing human ROR1-expressing murine TCL1 model of CLL and intraperitoneal dosing scheme for fully humanized huXBR1-402. Briefly, C57BL/6 mice were engrafted with 5 × 106 huROR1-TCL1 spleen-derived leukemic cells via tail vein injection. All mice were treated with 1 mg/kg of either huXBR1-402-G5-PNU, Tras-G5-PNU, or equal volumes of vehicle 3 times per week for 1 week after enrollment criteria were met (peripheral leukemia proportion was >5% of total white blood cells). (C) Weekly peripheral leukemia cell counts of a single mouse cohort as determined by flow cytometry. Mixed effects model was used to analyze differences with P value given at week 6. n = 4 mice per group. (D) Kaplan-Meier survival curve showing death of mice in each treatment group (vehicle, trastuzumab-PNU, unconjugated huXBR1-402, and huXBR1-402 PNU). Survival data were analyzed by using the log-rank test. n = 9 mice per group. Error bars indicate ± SE.
huXBR1-402-PNU suppressed leukemic growth and increased overall survival in models of aggressive human and murine ROR1+ leukemia. (A) 697 ROR1+ ALL disseminated leukemia model. Kaplan-Meier survival curve showing death of mice in each treatment group (trastuzumab-G5-PNU [Tras-G5-PNU] and XBR1-402-G5-PNU). Mice were treated intravenously with 1 mg/kg of drug on days 7 and 14 after implantation. Survival data were analyzed with log-rank test. n = 6 mice per group. (B) Schematic showing the experimental design for establishing human ROR1-expressing murine TCL1 model of CLL and intraperitoneal dosing scheme for fully humanized huXBR1-402. Briefly, C57BL/6 mice were engrafted with 5 × 106 huROR1-TCL1 spleen-derived leukemic cells via tail vein injection. All mice were treated with 1 mg/kg of either huXBR1-402-G5-PNU, Tras-G5-PNU, or equal volumes of vehicle 3 times per week for 1 week after enrollment criteria were met (peripheral leukemia proportion was >5% of total white blood cells). (C) Weekly peripheral leukemia cell counts of a single mouse cohort as determined by flow cytometry. Mixed effects model was used to analyze differences with P value given at week 6. n = 4 mice per group. (D) Kaplan-Meier survival curve showing death of mice in each treatment group (vehicle, trastuzumab-PNU, unconjugated huXBR1-402, and huXBR1-402 PNU). Survival data were analyzed by using the log-rank test. n = 9 mice per group. Error bars indicate ± SE.
We then evaluated the fully humanized huXBR1-402-G5-PNU's effects and dosing in an immune competent model of murine leukemia. A double transgenic huROR1-TCL1 leukemia mouse line was used that expresses human ROR1 on the surface of leukemic B cells. In addition to expressing human ROR1, the huROR1-TCL1 adoptive transfer mouse model is useful for evaluating the efficacy of huXBR1-402-G5-PNU as it models an aggressive CLL-like disease with a proliferative splenic niche (Figure 3B; supplemental Figure 6).12,32 Although anthracyclines are no longer routinely used for first-line CLL therapy, the immune-competent huROR1-TCL1 engrafted mice can be used to model aggressive, human ROR1+ lymphomas with proliferative niches. Treatment with 3 doses of huXBR1-402-G5-PNU (1 mg/kg) for 1 week postenrollment showed disease stabilization over a prolonged period of 3 weeks after the last dose and increased survival by 3 weeks in the huXBR1-402-G5-PNU treatment group. No toxicities were observed in either the trastuzumab-G5-PNU (tras-G5-PNU) or the huXBR1-402-G5-PNU treatment group (Figure 3C-D; supplemental Figure 7).
In vivo EdU cell cycle analysis confirmed our in vitro observations that huXBR1-402-G5-PNU targets proliferating leukemia cells, as evidenced by a decrease in the numbers of EdU+ CD19+/CD5+ leukemia cells in the spleens of huXBR1-402-G5-PNU–treated mice compared with vehicle and unconjugated huXBR1-402 control mice (Figure 4A). Although no significant differences were observed between huXBR1-402-G5-PNU and its isotype control during the short 24-hour pulse, this is presumably due to limited replication of leukemic cells within this short time period. Despite this, the splenic mass of treated mice was significantly decreased in animals given huXBR1-402-G5-PNU compared with all control mice (Figure 4B). Short-term follow-up of peripheral blood revealed almost double numbers of circulating tumor cells 4 days before euthanasia in vehicle and trastuzumab-G5-PNU–treated control animals compared with huXBR1-402-G5-PNU–treated animals (Figure 4C). No significant difference was noted compared with the unconjugated huXBR1-402 antibody, indicating a potential involvement of effector cell–mediated activity of huXBR1-402, as was observed in vitro.
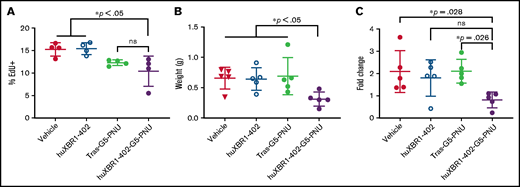
HuXBR1-402 targets proliferating leukemia cells in vivo. Briefly, 5 × 106 hROR1+ TLC1 murine CLL splenocytes were engrafted via tail vein injection into 8-week-old C57BL/6 mice. After establishment of leukemia (peripheral leukemia proportion is >5% of total white blood cells), mice were treated 3 times a week for 1 week with 1 mg/kg huXBR1-402-PNU or relevant controls. Three days after the last treatment, mice were pulsed with EdU (50 µg/kg) and killed 24 hours later to obtain the following data: numbers of EdU+ CD19+/CD5+ leukemia B cells (A) from splenocytes, obtained with flow cytometry (n = 4 mice per group); spleen weight (grams) (B); and fold change (C) in the numbers of peripheral blood leukemia cells. Mice were bled 4 days before euthanasia, and blood was again collected at time of euthanasia. Fold change in leukemia numbers are calculated by (peripheral leukemia cell count at death)/(peripheral leukemia cell count 4 days before). Panels B and C used n = 5 mice per group. Statistical significance for multiple comparisons was analyzed via one-way analysis of variance with Tukey’s post hoc test. ns, not significant; Tras-G5-PNU, trastuzumab-G5-PNU. Error bars indicate ± SE.
HuXBR1-402 targets proliferating leukemia cells in vivo. Briefly, 5 × 106 hROR1+ TLC1 murine CLL splenocytes were engrafted via tail vein injection into 8-week-old C57BL/6 mice. After establishment of leukemia (peripheral leukemia proportion is >5% of total white blood cells), mice were treated 3 times a week for 1 week with 1 mg/kg huXBR1-402-PNU or relevant controls. Three days after the last treatment, mice were pulsed with EdU (50 µg/kg) and killed 24 hours later to obtain the following data: numbers of EdU+ CD19+/CD5+ leukemia B cells (A) from splenocytes, obtained with flow cytometry (n = 4 mice per group); spleen weight (grams) (B); and fold change (C) in the numbers of peripheral blood leukemia cells. Mice were bled 4 days before euthanasia, and blood was again collected at time of euthanasia. Fold change in leukemia numbers are calculated by (peripheral leukemia cell count at death)/(peripheral leukemia cell count 4 days before). Panels B and C used n = 5 mice per group. Statistical significance for multiple comparisons was analyzed via one-way analysis of variance with Tukey’s post hoc test. ns, not significant; Tras-G5-PNU, trastuzumab-G5-PNU. Error bars indicate ± SE.
huXBR1-402-G5-PNU synergizes with ABT-199 (venetoclax) in a BCL2-dependent manner
Pre–B-ALL and MCL are tumors characterized by both actively proliferating and antiapoptotic lymphocytes.33-35 The antiapoptotic protein BCL2 is believed to mediate tumor cell therapy resistance and survival, and it is found at high levels in both ALL and MCL.36-38 Venetoclax, a BH3 mimetic, inhibits the antiapoptotic actions of BCL2 and is currently in trials for both MCL and pre–B-ALL after showing promising preclinical efficacy.39-42 Moreover, studies in triple-negative breast cancer have shown that BCL2 levels are an important indicator for response to anthracycline therapy, with tumors with lower BCL2 expression more responsive to anthracyclines.43,44 We therefore hypothesized that a combination of huXBR1-402-G5-PNU and venetoclax would show synergy and could be used to simultaneously target both proliferative and antiapoptotic tumor niches in ROR1+ tumors.
We tested combinations of serially diluted huXBR1-402-G5-PNU and venetoclax in ROR1+ ALL and MCL cell lines. As expected, strong Loewe additivity scores were observed for combinations of huXBR1-402-G5-PNU and venetoclax treatment in 697 and Mino cell lines (Figure 5A). No synergy was seen with the Tras-PNU isotype control. Of particular interest, among the different ROR1+ lines, 697 and Mino cells were also the most sensitive to venetoclax (50% effective concentration of 10.4 nM and 152 nM, respectively) (Figure 5D; supplemental Figure 8A). In contrast, venetoclax-resistant ROR1+cell lines such as JeKo-1, and HG3 cells showed no evidence of synergy at the concentrations tested (supplemental Figure 8B-C). We therefore postulated that synergy between the 2 compounds must be in part dependent on BCL2 expression. Not surprisingly, overexpression of BCL2 in 697 cells resulted in venetoclax resistance but also abrogated the cytotoxicity of huXBR1-402-G5-PNU treatment (Figure 5B-D; supplemental Figure 9). Unlike the low BCL2-expressing parental cell line, combination therapy with huXBR1-402-G5-PNU and venetoclax do not show synergy (Figure 5E). These findings warrant further clinical investigations into synergy between huXBR1-402-G5-PNU and venetoclax.
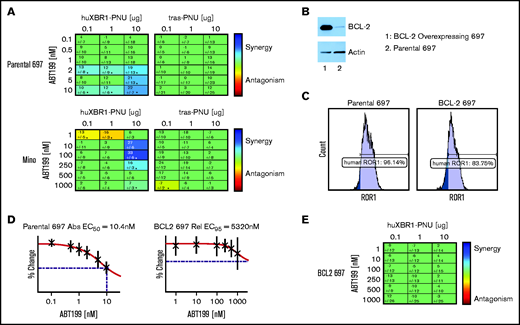
HuXBR1-402-G5-PNU synergizes with the BCL2 inhibitor venetoclax. (A) Loewe additivity synergy matrix with venetoclax and huXBR1-402-G5-PNU (left panel) or trastuzumab-G5-PNU (Tras-G5-PNU) (right panel) in the venetoclax-sensitive ROR1+ cell lines, 697 (top row) and Mino (bottom row). Synergy matrices display calculated Loewe additivity scores. Blue indicates doses in which synergy is found, and red indicates antagonism; green represents doses in which neither synergy nor antagonism is found. (B) BCL2 protein expression in ectopic BCL2 overexpressing (lane 1) and parental 697 (lane 2) (top) and corresponding actin protein expression on bottom. (C) Flow cytometry analysis of surface ROR1 expression comparison between parental (left) and BCL2 overexpression 697 cells (right). (D) Fifty percent effective concentration (EC50) of venetoclax treated parental (left) and BCL2-overexpressing (right). (E) Synergy matrix of combination venetoclax and huXBR1-402-G5-PNU in BCL2 overexpressing 697 cells. All experiments were completed in at least 3 independent experiments. Error bars indicate ± SE.
HuXBR1-402-G5-PNU synergizes with the BCL2 inhibitor venetoclax. (A) Loewe additivity synergy matrix with venetoclax and huXBR1-402-G5-PNU (left panel) or trastuzumab-G5-PNU (Tras-G5-PNU) (right panel) in the venetoclax-sensitive ROR1+ cell lines, 697 (top row) and Mino (bottom row). Synergy matrices display calculated Loewe additivity scores. Blue indicates doses in which synergy is found, and red indicates antagonism; green represents doses in which neither synergy nor antagonism is found. (B) BCL2 protein expression in ectopic BCL2 overexpressing (lane 1) and parental 697 (lane 2) (top) and corresponding actin protein expression on bottom. (C) Flow cytometry analysis of surface ROR1 expression comparison between parental (left) and BCL2 overexpression 697 cells (right). (D) Fifty percent effective concentration (EC50) of venetoclax treated parental (left) and BCL2-overexpressing (right). (E) Synergy matrix of combination venetoclax and huXBR1-402-G5-PNU in BCL2 overexpressing 697 cells. All experiments were completed in at least 3 independent experiments. Error bars indicate ± SE.
Discussion
Tumor antigen-targeted ADCs can greatly reduce systemic exposure to the toxic side effects of potent compounds, making them an attractive alternative to systemic chemotherapy. In the current study, we show the promising preclinical efficacy of the first-in-class anti–human-ROR1 ADC huXBR1-402-G5-PNU in controlling the proliferation of aggressive, ROR1+ hematologic malignancies in vitro and in vivo. The mechanism of huXBR1-402-G5-PNU–mediated cytotoxicity was shown to be BCL2 dependent; accordingly, it strongly synergizes with venetoclax in BCL2 inhibition–sensitive tumors such as ALL and subsets of MCL. Our results show the potential clinical application of a ROR1-targeted ADC, huXBR1-402-G5-PNU, for ROR1+ ALL and MCL.
ADCs such as huXBR1-402-G5-PNU allow us to harness the specificity of the ROR1- targeting antibody to deliver potent toxins without causing off-target toxicities. The safety of anti-ROR1 therapies has been established in several early clinical trials despite studies that show ROR1 expression on normal tissues, including the parathyroid, pancreas, and gut.6,16,17 Results of a phase 1 clinical trial with cirmtuzumab in CLL found excellent tolerability, with no dose-limiting toxicities and no high-grade adverse events after 4 biweekly doses of up to 20 mg/kg, suggesting that healthy tissues generally do not lead to on-target toxicities (potentially due to a low antigen expression level).18 More importantly, results of a phase 1 clinical trial with ROR1-specific CAR-T cells based off of the ROR1-2A2 antibody epitope in triple-negative breast and non–small cell lung cancer found no dose-limiting toxicities after infusions of up to 1 × 107 ROR1 CAR-T cells.16 Because CAR-T cells can target cells with ultra-low antigen density, the results of this trial underscore the general safety of ROR1-targeted therapies despite reports of low antigen expression on normal tissues.45 Our data and previously published studies of the parental rabbit clone establish that huXBR1-402 binds to the immunoglobulin and membrane distal-like/Frizzled portion of ROR1 that overlaps with the epitope of the anti-ROR1 CAR-T (Figure 1A), indicating that huXBR1-402-G5-PNU will likely also have limited off-target effects when used in patients.23 Accordingly, because normal tissue expression patterns of ROR1 are similar between humans and rhesus, rhesus models may be used in the future to assess the off-target effects of huXBR1-402-G5-PNU.46
Anthracyclines, primarily daunorubicin and doxorubicin, form the backbone of current MCL and ALL therapies due to their effectiveness in inhibiting DNA repair and replication in actively dividing cells.47-49 However, anthracycline therapy leads to multiple short- and long-term sequelae, including cardiotoxicity and eventual heart failure.50 By linking PNU, an anthracycline payload >2000 times more potent than daunorubicin, to a tumor-targeting ROR1 antibody, the risk of systemic side effects can be decreased and the effects of therapy concentrated on the tumor cells.51 Furthermore, recent studies have shown that PNU-159682–based payloads can enhance antitumor immunity through activation of a CD8 T cell–driven immune response.25 Although no toxicities were observed in our murine models, further studies in primates or humanized mice will be required to assess the stability, off-target toxicities, and immune-activating effects of huXBR1-402-G5-PNU.
The antiapoptotic protein BCL2 has increased activity and is overexpressed in both ALL and subsets of MCL.40,52-54 Venetoclax, a BCL2 inhibitor, has shown promising results in early clinical trials for both refractory ALL and MCL.42,55 Using human ALL and MCL tumor cell lines, our experiments show strong BCL2-dependent synergy between huXBR1-402-G5-PNU and venetoclax, suggesting that combined therapy should be further investigated in the clinic setting. Synergy between anthracyclines and BCL2 inhibitors is not surprising given that high BCL2 expression in early pre–B cells renders ALL cells more resistant to apoptosis induced by γ-irradiation–mediated DNA damage.56 In a myeloid tumor and hepatoblastoma cell line (HL60), inhibition of BCL2 has been shown to overcome resistance to doxorubicin-DNA adducts.57 Although the synergistic effects of huXBR1-402-G5-PNU and venetoclax can be seen in cells with low levels of BCL2, responses to huXBR1-402-G5-PNU were eliminated when BCL2 was increased in previously sensitive 697 cells. In line with this observation, in triple-negative breast cancer, BCL2 overexpression is correlated to poor responses to adjuvant anthracycline-based regiments.58 One critical factor that will need to be addressed in subsequent studies is identifying the dose response of huXBR1-402-G5-PNU at different expression levels of BLC2 and ways to overcome this BCL2-mediated resistance as this will determine the effectiveness and response to therapy in individual patients. However, given the results of promising preclinical trials for venetoclax in MCL and pre–B-ALL, synergy between huXBR1-402-G5-PNU and BCL2 inhibitors should be further characterized.
Although huXBR1-402-G5-PNU–mediated G2/M cell cycle arrest targets rapidly proliferating MCL, pre–B-ALL cells, and murine CLL-like ROR1+ tumor cells, it is not effective toward nonproliferative tumors such as ROR1+ CLL (Figure 2E). This is likely due to the cell cycle dependency of the anthracycline-based PNU payload. However, huXBR1-402-G5-PNU may still be effective in targeting proliferative tumor niches within CLL. Although the majority of circulating CLL cells in patients are nonproliferative, in vivo measurements of the birth rate of CLL cells and lymph node biopsy specimens from patients show the existence of proliferation centers in lymphoid organs that can drive disease progression.30,59,60 Measurements of in vivo proliferation showed significant decreases in the size of both splenic and circulating leukemia compartments in the huROR1-TCL1 leukemia model, indicating that huXBR1-402-G5-PNU is able to penetrate and target proliferative tissue niches. It is likely that combination therapy with apoptosis-promoting drugs such as BCL2 inhibitors may be more effective in controlling disease. Establishing the proliferative index of tumors will be critical for the clinical application of huXBR1-402-G5-PNU. In addition, relatively quiescent subpopulations of tumor-initiating cells may also not be effectively targeted with huXBR1-402-G5-PNU. Alternative payloads that do not depend on cell cycling status are currently being investigated for CLL and other nonproliferative ROR1+ tumors.
In summary, we show that huXBR1-402-G5-PNU is an effective therapeutic agent for ROR1+ hematologic malignancies. Our studies show that huXBR1-402-G5-PNU effectively represses leukemia growth in vitro and in vivo. Taken together with our experiments describing strong synergistic effects between huXBR1-402-G5-PNU and venetoclax, ROR1-targeted therapies can be potentially leveraged in combination therapies for multiple ROR1+ leukemia and lymphomas. Overall, our data show that huXBR1-402-G5-PNU is a promising modality for immunotherapy, with potential for broader applications in multiple solid and hematologic malignancies.
Acknowledgments
The authors are grateful to the patients who contributed to these studies and to David Lucas and Donna Bucci of the OSU Comprehensive Cancer Center Leukemia Tissue Bank Shared Resource (P30CA016058).
This work was supported by the National Institutes of Health, National Cancer Institute (R01-CA197844-01), the Pelotonia Idea Award, NBE-Therapeutics Ltd., and Robert J. Anthony Leukemia Fund. E.H. is funded by the National Cancer Institute F30 Grant (F30-CA225070-02) and by the Pelotonia Graduate Fellowship.
Authorship
Contribution: E.Y.H. designed, conducted and analyzed experiments, and wrote the manuscript; P.D. designed, conducted and analyzed experiments, and edited the manuscript; C. Chiang, C. Cheney, S.G., J.N., S.E., A.M.V., K.Z., E.W., R.M., F.F., H.P., C.R., L.W., M.L., and R.R.B. conducted and analyzed experiments, and edited the manuscript; X.M. conducted the statistical analyses for all experiments; and U.G., J.C.B., and N.M. conceptualized and designed the experiments, and edited the manuscript and all authors analyzed the data, reviewed the manuscript, and agree with submission for publication.
Conflict-of-interest disclosure: L.W., R.R.B., and U.G. are employed by NBE-Therapeutics Ltd. C.R. previously served on the Scientific Advisory Board of NBE-Therapeutics Ltd. C.R., H.P., R.R.B., U.G., and L.W. are named inventors on patents and patent applications that claim huXBR1-402 and are owned by The Scripps Research Institute and NBE-Therapeutics Ltd. The remaining authors declare no competing financial interests.
Correspondence: Natarajan Muthusamy, Division of Hematology, Department of Internal Medicine, The Ohio State University Comprehensive Cancer Center, 455E OSUCCC Bldg, 410 West 12th Ave, Columbus, OH 43210; e-mail: raj.muthusamy@osumc.edu.

