

Key Points
Red cells with a gain-of-function mutation in the calmodulin-binding domain of the KCNN4 K+ channel show increased intracellular [Ca2+].
Channel interactions (KCNN4, Piezo1, CaV2.1) contribute to the increased Ca2+ content in red cells of Gárdos channelopathy patients.

Abstract
In patients with Gárdos channelopathy (p.R352H), an increased concentration of intracellular Ca2+ was previously reported. This is a surprising finding because the Gárdos channel (KCa3.1) is a K+ channel. Here, we confirm the increased intracellular Ca2+ for patients with the KCa3.1 mutation p.S314P. Furthermore, we provide the concept of KCa3.1 activity resulting in a flickering of red blood cell (RBC) membranepotential, which activates the CaV2.1 channel allowing Ca2+ to enter the RBC. Activity of the nonselective cation channel Piezo1 modulates the aforementioned interplay in away that a closed Piezo1 is in favor of the KCa3.1-CaV2.1 interaction. In contrast, Piezo1 openings compromise the membrane potential flickering, thus limiting the activity of CaV2.1. With the compound NS309, we mimic a gain-of-function mutation of KCa3.1. Assessing the RBC Ca2+ response by Fluo-4–based flow cytometry and by measuring the membrane potential using the Macey-Bennekou-Egée method, we provide data that support the concept of the KCa3.1/CaV2.1/Piezo1 interplay as a partial explanation for an increased number of high Ca2+ RBCs. With the pharmacological inhibition of KCa3.1 (TRAM34 and Senicapoc), CaV2.1 (ω-agatoxin TK), and Piezo1 (GsMTx-4), we could project the NS309 behavior of healthy RBCs to the RBCs of Gárdos channelopathy patients.
Introduction
The Gárdos channel (hSK4, KCa3.1, KCNN4) is a Ca2+-activated K+ channel and was among the first ion channels described by the patch-clamp technique.1 In red blood cells (RBCs), the Gárdos channel was initially related to volume homeostasis and cell death.2,3 Only recently was its importance recognized for RBCs passing capillaries or constrictions.4-6 Gain-of-function mutations in the Gárdos channel have been associated with hereditary hemolytic anemia,7-13 either referred to as hereditary stomatocytosis caused by a Gárdos channel mutation7-9,11,12 or more directly as Gárdos channelopathy.10,12,13 For one of the identified mutations (p.R352H), increased intracellular Ca2+ content was reported10; for other genetic variants, it was not investigated. Because the Gárdos channel is a K+ channel, the mechanism of the Ca2+ increase is completely elusive. Here, experimental evidence is provided for the molecular interactions relating Gárdos channel activity to increased intracellular-free Ca2+, a key player in accelerated RBC damage and development of anemia.14
Methods
RBC collection
The blood sampling and all investigations were performed in accordance with the Helsinki Declaration of 1975. Patients and healthy donors had given their consent to give blood samples for research purposes. The study was approved by the Ethical Committee at Foundation IRCCS Ca’ Granda Ospedale Maggiore of Milan and the Ärztekammer des Saarlandes (approval #51/18). The patients are described in separate previous reports.10,13 Blood samples were taken in Li-heparin tubes and shipped overnight from Milan and Padua (Italy) to Homburg (Germany).
Imaging and flow cytometry
Ca2+ imaging was performed as previously described15 and confocal images were recorded on a TCS SP5 (Leica, Germany) as outlined previously.16
For flow cytometry measurements, 30 000 RBCs loaded with Fluo-4 were analyzed with the flow cytometer as previously described.14,15 The percentage of responding RBCs is the statistically analyzed parameter.
Solutions, chemicals, and further procedures are detailed in supplemental Materials and methods.
Membrane potential measurements
The membrane potential measurements of a RBC population were performed according to a method initially described by Macey et al,17 further developed and applied by Poul Bennekou (ie, Baunbæk and Bennekou18), and kept alive by the laboratory of Stéphane Egée.13 Therefore, we refer to it as the Macey-Bennekou-Egée (MBE) method. A detailed description is provided in supplemental Materials and methods.
Results and discussion
RBCs from a patient carrying the KCNN4 p.S314P variant13 were tested for the free intracellular Ca2+ concentration, which was found significantly increased (Figure 1A) in agreement with a previous report in the p.R352H variant.10 Although such an increase in intracellular Ca2+ has been proposed as a common component of the mechanisms causing anemia,14 the causal link to a mutation in the Gárdos channel, which is a K+ channel, remains unclear. To this end, reference is made to a concept of the role of voltage-activated channels in nonexcitable cells19: it has been shown that Gárdos channel activation leads to a hyperpolarization of the RBC membrane from approximately −12 mV to −60 mV.13,18 Because the number of Gárdos channels in RBCs is very low (1-5 copies per cell in 75% of the cells20,21), and due to the stochastic behavior of channel openings, one can assume that, upon Gárdos channel activation, the membrane potential flickers (ie, jumps between −12 mV and −60 mV).19 This is the necessary condition to activate voltage-gated Ca2+ channels, such as CaV2.1, which was reported to be abundant in RBCs.22 The activation of the CaV2.1 channel would result in increased intracellular Ca2+. A scheme of this concept in the context of the gain-of-function Gárdos channel mutations is shown in Figure 1B.
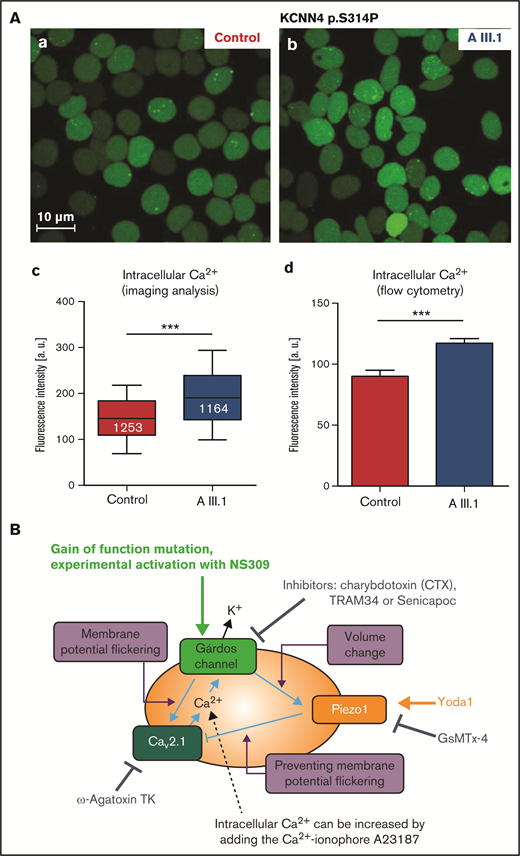
Ca2+ increase in RBCs with Gárdos channel mutations. (A) Ca2+ increase in RBCs of a patient with the p.S314P mutation (AIII.1 in Fermo et al13) in comparison with healthy control RBCs. (a-b) Representative fluorescence images of RBCs stained with Fluo-4; scale bar, 10 μm. (c) Statistical analysis of fluorescent images as depicted in subpanel a. The white numbers refer to the numbers of analyzed cells. (d) Analysis of flow cytometric measurements of Fluo-4–stained RBCs. Statistical differences were checked with a Student t test; *** P < .001. (B) Mechanistic hypothesis regarding how Gárdos channel activity may trigger an increase in intracellular RBC Ca2+ concentration. The membrane potential changes induced by the opening of the Gárdos channel (cp. explanation in "Results and discussion") activates the voltage-gated Ca2+ channel CaV2.1, possibly modulated by the mechanosensitive Piezo1 channel. If Piezo1 is blocked, the Gárdos channel–induced opening of CaV2.1 would be maximal. In contrast, Piezo1 activity, due to the nonselective cation permeability of the channel, would “dissipate” the Gárdos channel–induced hyperpolarization. As a consequence, CaV2.1 activity would be reduced. The scheme contains annotations for the experimental manipulations of the molecular players. An arrow refers to activation and an anchor to inhibition of the transport protein.
Ca2+ increase in RBCs with Gárdos channel mutations. (A) Ca2+ increase in RBCs of a patient with the p.S314P mutation (AIII.1 in Fermo et al13) in comparison with healthy control RBCs. (a-b) Representative fluorescence images of RBCs stained with Fluo-4; scale bar, 10 μm. (c) Statistical analysis of fluorescent images as depicted in subpanel a. The white numbers refer to the numbers of analyzed cells. (d) Analysis of flow cytometric measurements of Fluo-4–stained RBCs. Statistical differences were checked with a Student t test; *** P < .001. (B) Mechanistic hypothesis regarding how Gárdos channel activity may trigger an increase in intracellular RBC Ca2+ concentration. The membrane potential changes induced by the opening of the Gárdos channel (cp. explanation in "Results and discussion") activates the voltage-gated Ca2+ channel CaV2.1, possibly modulated by the mechanosensitive Piezo1 channel. If Piezo1 is blocked, the Gárdos channel–induced opening of CaV2.1 would be maximal. In contrast, Piezo1 activity, due to the nonselective cation permeability of the channel, would “dissipate” the Gárdos channel–induced hyperpolarization. As a consequence, CaV2.1 activity would be reduced. The scheme contains annotations for the experimental manipulations of the molecular players. An arrow refers to activation and an anchor to inhibition of the transport protein.
To experimentally test this model, we examined the effect of NS309, a compound able to increase the open probability of the Gárdos channel,18,23 independent of intracellular Ca2+; experiments with RBCs from healthy donors are shown in Figure 2A. NS309-induced Ca2+ entry is at least partly triggered by Gárdos channel activity as application of 2 different Gárdos channel blockers, charybdotoxin (CTX) and TRAM34, reveal (Figure 2Ab). Both blockers (CTX and TRAM34) alone had no influence on the number of high Ca2+ cells (supplemental Figure 1). In a new set of measurements, CaV2.1 and Piezo1 channel blockers ω-agatoxin TK and GsMTx-4 were applied, respectively. ω-agatoxin TK is a specific channel blocker,24 whereas GsMTx-4 blocks all kinds of mechanosensitive channels including Piezo1.25 The block of the CaV2.1 channel with ω-agatoxin TK reduces NS309-induced Ca2+ entry, whereas the block of Piezo1 with GsMTx-4 increases the number of high Ca2+ cells (Figure 2Ac). Both blockers (ω-agatoxin TK and GsMTx-4) alone had no influence on the number of high Ca2+ cells (supplemental Figure 1). Although the reduction of the number of high Ca2+ cells by CaV2.1 channel blocker ω-agatoxin TK looks obvious, the increase caused by the application of GsMTx-4 seems a paradox. However, referring to the concept outlined in Figure 1B, the inhibition of the nonselective cation channel Piezo1 is in favor of the flickering membrane potential: if the nonselective cation channel Piezo1 is in an open state, it depolarizes the RBC and thus disables the function of CaV2.1. To test this interpretation, we performed membrane potential measurements using the MBE method (Figure 2Ad), showing the hyperpolarization when the Gárdos channel is active and following depolarization when Piezo1 gets activated. In a previous article, the dependence of NS309-induced Gárdos channel activation on external Ca2+ concentration was shown,18 which we could confirm in terms of the induced membrane potential change (Figure 2Ae). Furthermore, our probe of this Ca2+ dependence for the percentage of RBCs responding with an increased Ca2+ concentration indicated a bell-shaped relation (Figure 2Af). Thus, the concept of the interplay of the Gárdos channel with CaV2.1 was further supported: when the open probability of the Gárdos channel is steadily increased, a condition is reached when the channels are predominantly open. This in turn results in a decrease of membrane potential flickering and hence less activity of CaV2.1 and a lower number of high Ca2+ cells.
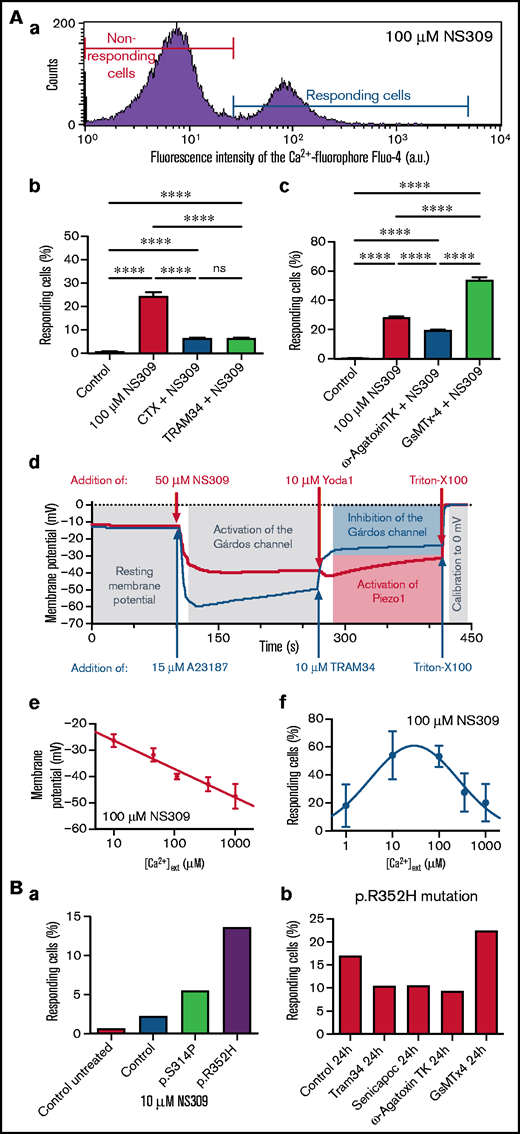
Experimental evidence for a mechanistic explanation. (A) Investigation of healthy RBCs to examine interactions between the Gárdos channel and other RBC ion channels. (a) Representative histogram from a flow cytometry measurement of RBCs stained with Fluo-4 and stimulated with 100 μM NS309. The red and the blue lines mark the populations of nonresponding and responding RBCs, respectively. (b) Number of RBCs responding with a high Ca2+ content as outlined in subpanel a at a concentration of 100 μM NS309 (left column) and the effect (preincubation) of the Gárdos channel inhibitors CTX (0.1 μM) and TRAM34 (1 μM) on NS309 (100 μM)-stimulated RBCs (right columns). (c) A new set of experiments compares the action (preincubation) of ω-Agatoxin TK (1 μM), a specific inhibitor of CaV2.1, and GsMTx-4 (2 μM), a toxin inhibiting the mechanosensitive channel Piezo1, on NS309 (100 μM)-stimulated RBCs. (b-c) All measurements are performed on at least 4 different donors. Plotted are mean values and the standard error of mean. Statistical differences were checked with a 1-way analysis of variance (ANOVA) and the Tukey multiple comparisons test. **** P < .0001; ns, not significant (P > .05). (d) Monitoring the membrane potential in a population of RBCs by the MBE method. The blue curve starts at the resting membrane potential (−12 mV). Addition of 15 μM A23187 leads to Ca2+ entry and the consecutive full activation of the Gárdos channel resulting in a hyperpolarization of −60 mV. Addition of TRAM34 inhibits the Gárdos channel resulting in a depolarization. The red curve also starts with the resting membrane potential and addition of 50 μM NS309 leads to the activation of the Gárdos channel, also resulting in a hyperpolarization but to a lesser extent as for the blue curve. Because we monitor the membrane potential in a cell population of >108 RBCs, it is impossible to see a membrane potential flickering (compare "Results and discussion") that may happen in individual cells. The hyperpolarization to only −40 mV is therefore caused by, to a lesser extent, Gárdos channel activation, that is, a composition of cells with open and closed Gárdos channels. For both experiments, Triton-X 100 is used to calibrate for 0 mV. The curves present the mean of a triplicate measurement of a healthy donor and are a representative of 3 different donors. (e) Membrane potential upon 100 μM NS309 stimulation in dependence of the extracellular Ca2+ concentration. (f) Percentage of high Ca2+ RBCs upon 100 μM NS309 stimulation in dependence of the external Ca2+concentration. The corresponding histograms are provided in supplemental Figure 2. The bell-shaped curve can be explained by the Gárdos channel–CaV2.1 interaction (see main text). (e-f) All measurements were performed on 3 different donors. Plotted are mean values and the standard error of mean. (B) Testing the principle investigated in panel A on RBCs of patients carrying a Gárdos channel mutation. (a) Differential response (number of cells responding with increased intracellular Ca2+) of healthy and KCNN4 p.S314P- and p.R352H-mutated RBCs on stimulation with 10 μM NS309. (b) p.R352H-mutated RBCs were incubated for 24 hours with the Gárdos channel inhibitors Senicapoc (5 μM) and TRAM34 (1 μM), the CaV2.1 inhibitor ω-Agatoxin TK (1 μM), and the Piezo1 inhibitor GsMTx-4 (2 μM). The number of cells responding with increased intracellular Ca2+ is shown.
Experimental evidence for a mechanistic explanation. (A) Investigation of healthy RBCs to examine interactions between the Gárdos channel and other RBC ion channels. (a) Representative histogram from a flow cytometry measurement of RBCs stained with Fluo-4 and stimulated with 100 μM NS309. The red and the blue lines mark the populations of nonresponding and responding RBCs, respectively. (b) Number of RBCs responding with a high Ca2+ content as outlined in subpanel a at a concentration of 100 μM NS309 (left column) and the effect (preincubation) of the Gárdos channel inhibitors CTX (0.1 μM) and TRAM34 (1 μM) on NS309 (100 μM)-stimulated RBCs (right columns). (c) A new set of experiments compares the action (preincubation) of ω-Agatoxin TK (1 μM), a specific inhibitor of CaV2.1, and GsMTx-4 (2 μM), a toxin inhibiting the mechanosensitive channel Piezo1, on NS309 (100 μM)-stimulated RBCs. (b-c) All measurements are performed on at least 4 different donors. Plotted are mean values and the standard error of mean. Statistical differences were checked with a 1-way analysis of variance (ANOVA) and the Tukey multiple comparisons test. **** P < .0001; ns, not significant (P > .05). (d) Monitoring the membrane potential in a population of RBCs by the MBE method. The blue curve starts at the resting membrane potential (−12 mV). Addition of 15 μM A23187 leads to Ca2+ entry and the consecutive full activation of the Gárdos channel resulting in a hyperpolarization of −60 mV. Addition of TRAM34 inhibits the Gárdos channel resulting in a depolarization. The red curve also starts with the resting membrane potential and addition of 50 μM NS309 leads to the activation of the Gárdos channel, also resulting in a hyperpolarization but to a lesser extent as for the blue curve. Because we monitor the membrane potential in a cell population of >108 RBCs, it is impossible to see a membrane potential flickering (compare "Results and discussion") that may happen in individual cells. The hyperpolarization to only −40 mV is therefore caused by, to a lesser extent, Gárdos channel activation, that is, a composition of cells with open and closed Gárdos channels. For both experiments, Triton-X 100 is used to calibrate for 0 mV. The curves present the mean of a triplicate measurement of a healthy donor and are a representative of 3 different donors. (e) Membrane potential upon 100 μM NS309 stimulation in dependence of the extracellular Ca2+ concentration. (f) Percentage of high Ca2+ RBCs upon 100 μM NS309 stimulation in dependence of the external Ca2+concentration. The corresponding histograms are provided in supplemental Figure 2. The bell-shaped curve can be explained by the Gárdos channel–CaV2.1 interaction (see main text). (e-f) All measurements were performed on 3 different donors. Plotted are mean values and the standard error of mean. (B) Testing the principle investigated in panel A on RBCs of patients carrying a Gárdos channel mutation. (a) Differential response (number of cells responding with increased intracellular Ca2+) of healthy and KCNN4 p.S314P- and p.R352H-mutated RBCs on stimulation with 10 μM NS309. (b) p.R352H-mutated RBCs were incubated for 24 hours with the Gárdos channel inhibitors Senicapoc (5 μM) and TRAM34 (1 μM), the CaV2.1 inhibitor ω-Agatoxin TK (1 μM), and the Piezo1 inhibitor GsMTx-4 (2 μM). The number of cells responding with increased intracellular Ca2+ is shown.
In Figure 2Ba, the different sensitivity of the Gárdos channel mutations towards application of NS309 initially reported by Fermo et al13 is shown and was also reflected in the number of cells responding with an increased Ca2+ concentration. We aimed to test whether the mechanism induced by stimulation with NS309 in healthy cells (Figure 2Ac) was also applicable to cells with the Gárdos channel mutation. NS309 increases the open probability of the Gárdos channel; gain-of-function mutations of the channel should lead to a similar result. Because the Gárdos channel gain-of-function mutation could not be “switched on” (as the Gárdos channel can with the application of NS309), the effect of the channel blockers was compared in a 24-hour “long-term” experiment. RBCs carrying the mutation p.R352H were incubated with the respective inhibitors for 24 hours and the number of RBCs with increased Ca2+ was measured (Figure 2Bb). Although statistical analysis was not available due to sample limitation, the result shown echoes the observation in the NS309-stimulated healthy cells (Figure 2Ac).
These data further support the concept of ion channel interplay being causal for the increased Ca2+ concentration in Gárdos channelopathy RBCs. The given interpretation for the increased Ca2+ in the RBCs of KCNN4-mutated patients may have evident bias because the channel inhibitors only block a fraction of the measured effect, both in the NS309 model (Figure 2A) as well as in the patient RBCs (Figure 2Bb). We present experimental indication of >2 direct ion channel interactions in RBCs that are otherwise rather typical for excitable cells. Further research is required to substantiate and extend the results presented in this stimulus report.
Acknowledgments
This work was supported by European Framework Horizon 2020 under grant agreement number 860436 (EVIDENCE) and by Fondazione IRCCS Ca’ Granda Policlinico Milano, project number RC2020 175/05.
Authorship
Contribution: L.K. and P.B. designed the study; E.F., P.B., and A.Z. performed patient diagnostic workup; L.K. and I.B. supervised the experiments; J.J., M.Q., L.H., X.W., and E.F. performed the experiments; R.C. followed-up with patients; L.K. drafted the manuscript; and all authors revised and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Lars Kaestner, Saarland University, Building E2_6, 66123 Saarbrücken, Germany; e-mail: lars_kaestner@me.com.

