

Key Points
Avelumab was well tolerated in previously treated cHL, consistent with its known safety profile.
ORR (95% confidence interval) was 41.9% (24.5%-60.9%) in all patients and 55.6% (21.2%-86.3%) in those who received prior allo-HSCT.

Abstract
The 9p24.1 chromosomal alteration in classical Hodgkin lymphoma (cHL) is associated with increased expression of programmed death ligand 1 (PD-L1)/PD-L2 and an immunosuppressive tumor microenvironment. Blockade of PD-L1/PD-1 interactions with avelumab (anti–PD-L1) is hypothesized to restore antitumor immunity. JAVELIN Hodgkins was a phase 1b, multiple-dose, open-label, randomized, parallel-arm trial of avelumab in patients with relapsed/refractory (R/R) cHL. Primary end points included avelumab target occupancy by dose/schedule in peripheral blood immune cells and pharmacokinetic parameters. Secondary end points included safety and antitumor activity. Four dose levels and 2 dosing schedules were investigated: 70, 350, and 500 mg administered every 2 weeks; 500 mg every 3 weeks; and 10 mg/kg every 2 weeks. Thirty-one patients with R/R cHL were randomized; 9 (29.0%) and 20 (64.5%) had received 3 or ≥4 prior anticancer treatments, respectively. Target occupancy of >90% was observed across all treatment arms, throughout the dosing interval. Avelumab pharmacokinetic data were similar to those previously reported. The most common treatment-related adverse events of any grade were infusion-related reaction (30.0%), nausea (20.0%), increased alanine aminotransferase and rash (16.7% each), and fatigue (13.3%). The objective response rate (ORR) in all randomized patients was 41.9%, with a complete response rate of 19.4%; ORR in those with prior allogeneic hematopoietic stem cell transplant (allo-HSCT) was 55.6%. Due to decreased use of allo-HSCT in patients with R/R cHL, the expansion phase enrolling post–allo-HSCT patients was terminated. Avelumab was tolerable and demonstrated antitumor activity in heavily pretreated patients with cHL, suggesting that PD-L1 blockade may be sufficient for therapeutic benefit in cHL. This trial was registered at www.clinicaltrials.gov as #NCT02603419.
Introduction
Classical Hodgkin lymphoma (cHL) is characterized by a heterogeneous tumor microenvironment that is rich in reactive immune cells and has few Hodgkin and Reed-Sternberg tumor cells.1 These tumor cells exhibit deregulated activity of signaling pathways that directly promote tumor cell survival, proliferation, and immune evasion. Genetic mutations and chromosomal copy-number alterations involving various tumor suppressors and proto-oncogenes result in constitutive activation of the antiapoptotic/prosurvival NF-κB and JAK-STAT signaling pathways.1-3 Among the various copy-number gains, amplification of chromosome 9p24.1 is frequently observed.4,5 The resulting amplicon contains the genes encoding JAK2 and the programmed death ligand 1 (PD-L1) and PD-L2 immune-checkpoint proteins, which results in their overexpression and contributes to the immunosuppressive tumor microenvironment.5
Binding of programmed cell death protein 1 (PD-1) on T cells by its ligands PD-L1 and PD-L2, which are expressed on tumor cells, suppresses effector T-cell function and promotes immune evasion.2 Clinical trials investigating the efficacy and safety of the anti–PD-1 antibodies nivolumab and pembrolizumab in patients with relapsed/refractory (R/R) cHL have shown durable responses in many patients,6-12 and both drugs are approved for this indication.13,14 These anti–PD-1 antibodies block both PD-1/PD-L1 and PD-1/PD-L2 interactions; however, it is unknown whether use of PD-L1 inhibitors, which inhibit the PD-1/PD-L1 interaction but leave the PD-1/PD-L2 interaction intact, is sufficient to achieve the therapeutic effect observed with anti–PD-1 antibodies in cHL. Avelumab is a human anti–PD-L1 immunoglobulin G1 monoclonal antibody that specifically inhibits PD-1/PD-L1 interactions,15 and induces innate effector function against tumor cells in vitro through its functional Fc portion.16,17 Avelumab monotherapy has shown antitumor activity and a manageable safety profile in Merkel cell, urothelial, and renal cancer carcinomas, and several other solid tumor subtypes.18-23
Here, we report results from the phase 1b JAVELIN Hodgkins study, which was the first trial to assess the pharmacokinetics (PK), efficacy, and safety of avelumab in patients with R/R cHL as well as the first to report on patients who received PD-L1 blockade following disease progression after allogeneic hematopoietic stem cell transplant (allo-HSCT).
Methods
Patients
JAVELIN Hodgkins was a phase 1b, open-label, multicenter clinical trial with a lead-in phase and a planned expansion phase that enrolled patients at 7 sites in the United States and western Europe. The lead-in phase was a multiple-dose, randomized, parallel-arm, PK, and pharmacodynamic study of avelumab as a single agent in adults with R/R cHL. An expansion phase based on the preliminary target occupancy (TO), safety, and efficacy results from the lead-in phase was planned but was terminated due to recruitment difficulties arising from rapidly evolving standards of care for this disease. Here, we present study design and results of the lead-in phase of JAVELIN Hodgkins and briefly describe results from 3 patients in the expansion phase.
Eligible patients had histologically confirmed cHL that relapsed following a prior autologous HSCT (auto-HSCT) or allo-HSCT or were ineligible for HSCT. Other key eligibility criteria included completion of prior therapy ≥28 days prior to randomization; ≥1 fluorodeoxyglucose positron emission tomography (PET)–avid measurable lesion >1.5 cm on a PET–computed tomography (PET-CT) scan as defined by the revised response criteria for malignant lymphoma;24 age ≥18 years; Eastern Cooperative Oncology Group performance status of 0 or 1; and adequate bone marrow, renal, and liver function. Reasons for exclusion included prior allo-HSCT performed <12 months prior to randomization, and, for patients who had received an allo-HSCT, grade 3 or 4 acute graft-versus-host disease (GVHD) at any time in the past as defined by the modified Seattle Glucksberg criteria (Consensus Conference on Acute GVHD Grading criteria).25 Patients with a history of chronic GVHD, as defined by the National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease,26 that persisted for >6 months and required systemic immunosuppression were excluded except for those who required ≤15 mg per day of oral prednisone or equivalent (must have discontinued ≤7 days prior to the first dose of study treatment). Patients who received immunosuppressive treatment of acute or chronic GVHD ≤3 months prior to randomization were also excluded except for those who required ≤15 mg per day of oral prednisone or equivalent that was discontinued >7 days prior to the first dose of study treatment. Patients who used immunosuppressive medication ≤7 days prior to randomization were excluded, with the exception of intranasal, inhaled, topical steroids or local steroid injections; systemic corticosteroids (at physiological doses ≤10 mg per day of prednisone or equivalent) for the treatment of adrenal insufficiency; and steroids as premedication for hypersensitivity reactions. Patients who received prior therapy with an anti–PD-1 or anti–PD-L1 antibody were not eligible, unless treatment was stopped >1 year prior to randomization and they had a documented prior response; those with a prior history of grade ≥3 anti–PD-1/PD-L1–related immune toxicity were also excluded.
All patients provided written informed consent. This study was approved by the relevant institutional review boards or ethics committees at all participating centers and was conducted in accordance with the principles of the Declaration of Helsinki. All authors had full access to all data, and the first author had final responsibility for the decision to submit the manuscript for publication.
Randomization, treatment, end points, and assessments
An interactive response technology system/interactive web response was used to randomize (1:1:1:1:1) ∼30 patients in parallel across 5 avelumab treatment groups, 4 receiving flat dosing (70 mg every 2 weeks, 350 mg every 2 weeks, 500 mg every 2 weeks, and 500 mg every 3 weeks) and 1 receiving dosing by body weight (10 mg/kg every 2 weeks). These regimens were chosen to explore the influence of different doses, dosing frequencies, and fixed vs body weight–based dosing on PK and the TO time course. A cycle was defined as the time from the day 1 dose to the next day 1 dose; if there were no treatment delays, a cycle consisted of 2 weeks for every-2-weeks and 3 weeks for every-3-weeks dosing regimens.
Avelumab was administered as a 1-hour IV infusion; premedication with an antihistamine and acetaminophen ∼30 to 60 minutes prior to each dose of avelumab was mandatory. Alternative premedication schema could be selected based on local treatment standards and guidelines, as appropriate. Avelumab dose reductions for toxicity management were not permitted, but the next cycle administration could be omitted due to persisting toxicity. In the event of evidence of radiological progression, patients could continue to receive avelumab if they were clinically stable and continued to experience clinical benefit. Patients who achieved a complete response (CR) or partial response (PR) continued on treatment until disease progression, refusal, unacceptable toxicity, or loss to follow-up or until the study was terminated by the sponsor, whichever occurred first. Patients with progressive disease (PD) continued treatment if they were clinically stable and if repeat imaging at 6 weeks or earlier showed CR, PR, or stable disease (SD) compared with baseline. If repeat imaging confirmed PD, treatment was discontinued.
The primary end points were percentage TO by dose/schedule in peripheral blood immune cells, including CD14+ monocytes and CD3+ T cells, and PK parameters of avelumab, including maximum observed plasma concentration, predose trough concentration during multiple dosing (Ctrough), and area under the curve. Secondary end points included safety, objective response per response criteria for malignant lymphoma24 according to investigator assessment, disease control (defined as best overall response [BOR] of CR, PR, or SD), time to tumor response, duration of response, and progression-free survival (PFS) according to investigator assessment. Tumor assessments included all known or suspected disease sites. Imaging included neck, chest, abdomen, and pelvis PET-CT scans at baseline and could have additionally included CT with contrast or magnetic resonance imaging if 1 of these 2 modalities (the same type of scan as baseline) was continued for disease assessments; an additional PET-CT scan was performed at 6 weeks and subsequently as clinically indicated. PET-CT scanning was conducted at screening, 6 weeks, and 12 weeks, and at 12-week intervals thereafter until documented disease progression regardless of initiation of subsequent anticancer therapy. Additional tumor assessments were conducted whenever disease progression was suspected (eg, symptomatic deterioration), at the end of treatment/withdrawal (if not done in the previous 6 weeks), and at follow-up visits.
Statistical analysis
No formal hypothesis testing was done in the lead-in phase of the study. PK data from the lead-in phase were used to enable the initial estimation of TO in each treatment group; based on historical data,15 with an observed mean TO of 90%, the corresponding 95% confidence interval (CI) would be 88.98% to 91.02%, assuming a standard deviation of 1.27%. PK end points were summarized descriptively, as data permitted. The objective response rate (ORR) was calculated along with 2-sided 95% CI using the Clopper-Pearson method. Duration of response and PFS were estimated using the Kaplan-Meier method, and the CI for the median was calculated according to the Brookmeyer and Crowley method. The CI for the Kaplan-Meier rate was calculated using log-log transformation with back transformation to a CI on the untransformed scale.
Biomarker analyses
PD-L1 TO.
Blood samples for PD-L1 TO were collected at cycle 1 day 1 prior to dosing; cycle 1 days 2, 7, and 14 (every 3 weeks only); cycle 2 days 1, 2, 7, and 14 (every 3 weeks only); cycle 3 day 1; cycle 4 day 1; and the end of treatment. The cell-surface expression of free PD-L1 was measured on peripheral blood CD14+ monocytes or CD3+ T cells by flow cytometry using a phycoerythrin-labeled anti–PD-L1 flow detection antibody. The percentage of PD-L1+ cells and mean fluorescence intensity were evaluated. Free PD-L1 levels as measured by binding of the phycoerythrin-labeled detection antibody, which is noncompetitive with avelumab, were reduced or absent postdosing due to the presence of prebound avelumab. Hence, the percentage of TO could be calculated from the degree of reduction in the detection of antibody binding postdose compared with predose.
PD-L1/PD-L2 FISH and PD-L1 immunohistochemistry.
The fluorescence in situ hybridization (FISH) assay, Empire Genomics PD-L1 (CD274) and PD-L2 (PDCD1LG2) combined with Abbott Molecular CEP9, was performed on formalin-fixed, paraffin-embedded (FFPE) human tissue to determine 9p24.1 alterations of polysomy, copy gain, amplification, and rearrangement of the PD-L1 and PD-L2 genes. FFPE tissue sections on slides were deparaffinized. Cellular DNA was denatured to single-stranded form and subsequently allowed to hybridize with the applied DNA probes. Following hybridization, the slides were counterstained with 4′,6 diamidino-2-phenylindole (blue), a DNA-specific stain that fluoresces blue. Hybridization of cells with Empire Genomics probes for PD-L1 (CD274; orange) and PD-L2 (PDCD1LG2; green) combined with the Abbott Molecular CEP9 (aqua) probe was viewed using a fluorescence microscope equipped with appropriate excitation and emission filters, allowing visualization of fluorescent signals (orange, green, and aqua). Normal cells demonstrate 2 immediately adjacent or fused (overlapping) orange/green (yellow) signals and 2 aqua signals (disomy). Abnormal cells with alterations of 9p24.1 demonstrate polysomy, copy gain, amplification (copy-number alterations), or rearrangement signal patterns. Tumor cell PD-L1 protein expression was detected in FFPE tissue sections using the Dako PD-L1 IHC 22C3 pharmDx assay (performed by Hematogenix Laboratory Services, LLC, Tinley Park, IL). PD-L1 positivity in tumor cells was scored as the proportion of viable tumor cells showing membranous PD-L1 staining and the proportion of tumor-associated immune cells showing PD-L1 staining.
Results
Patients and treatment
Thirty-one patients were randomized in the lead-in phase between 15 March and 29 November 2016. The avelumab dosage was 70 mg every 2 weeks in 6 patients, 350 mg every 2 weeks in 7 patients, 500 mg every 2 weeks in 6 patients, 500 mg every 3 weeks in 6 patients, and 10 mg/kg every 2 weeks in 6 patients. Thirty patients received study treatment (1 patient randomized to the 350 mg every-2-weeks group did not receive avelumab). Baseline characteristics of all randomized patients are shown in Table 1. Most patients were male (77.4%). The median age was 38.0 years (range, 22.0-81.0 years). All patients had received prior treatment with brentuximab vedotin. None had received prior treatment with an immune-checkpoint inhibitor. Four patients (12.9%) received prior auto-HSCT only (auto-HSCT subgroup). Three patients (9.7%) received prior allo-HSCT only, and 6 (19.4%) received both prior auto-HSCT and prior allo-HSCT. Patients from the latter 2 groups (allo-HSCT only and allo-HSCT plus auto-HSCT) are included in the allo-HSCT subgroup. All patients in the auto-HSCT subgroup received study treatment. Eight patients in the allo-HSCT subgroup received study treatment (the patient in the 350 mg every-2-weeks group who did not receive study treatment had a history of prior auto-HSCT and allo-HSCT).
Baseline patient characteristics
| Characteristic | All patients, N = 31, median (range) or n (%) | Avelumab treatment group, median (range) or n (%) | ||||
|---|---|---|---|---|---|---|
| 70 mg Q2W, n = 6 | 350 mg Q2W, n = 7 | 500 mg Q2W, n = 6 | 500 mg Q3W, n = 6 | 10 mg/kg Q2W, n = 6 | ||
| Age, y | 38.0 (22.0-81.0) | 57.0 (31.0-78.0) | 48.0 (30.0-71.0) | 36.5 (22.0-56.0) | 35.0 (25.0-78.0) | 31.5 (25.0-81.0) |
| Age group, y | ||||||
| <65 | 24 (77.4) | 5 (83.3) | 4 (57.1) | 6 (100.0) | 4 (66.7) | 5 (83.3) |
| ≥65 | 7 (22.6) | 1 (16.7) | 3 (42.9) | 0 | 2 (33.3) | 1 (16.7) |
| ECOG performance status | ||||||
| 0 | 14 (45.2) | 2 (33.3) | 4 (57.1) | 3 (50.0) | 3 (50.0) | 2 (33.3) |
| 1 | 17 (54.8) | 4 (66.7) | 3 (42.9) | 3 (50.0) | 3 (50.0) | 4 (66.7) |
| Sex | ||||||
| Male | 24 (77.4) | 3 (50.0) | 6 (85.7) | 5 (83.3) | 5 (83.3) | 5 (83.3) |
| Female | 7 (22.6) | 3 (50.0) | 1 (14.3) | 1 (16.7) | 1 (16.7) | 1 (16.7) |
| Geographic region | ||||||
| North America | 10 (32.2) | 2 (33.3) | 2 (28.6) | 2 (33.3) | 2 (33.3) | 2 (33.3) |
| Western Europe | 21 (67.7) | 4 (66.7) | 5 (71.4) | 4 (66.7) | 4 (66.7) | 4 (66.7) |
| No. of prior anticancer therapy regimens | ||||||
| 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | 2 (6.5) | 0 | 0 | 0 | 1 (16.7) | 1 (16.7) |
| 3 | 9 (29.0) | 2 (33.0) | 2 (28.6) | 1 (16.7) | 2 (33.3) | 2 (33.3) |
| ≥4 | 20 (64.5) | 4 (66.7) | 5 (71.4) | 5 (83.3) | 3 (50.0) | 3 (50.0) |
| Prior treatment with brentuximab vedotin | 31 (100.0) | 6 (100.0) | 7 (100.0) | 6 (100.0) | 6 (100.0) | 6 (100.0) |
| Patients with prior HSCT | 13 (41.9) | 2 (33.3) | 2 (28.6) | 4 (66.7) | 4 (66.7) | 1 (16.7) |
| Type of HSCT | ||||||
| Autologous only | 4 (12.9) | 0 | 0 | 2 (33.3) | 2 (33.3) | 0 |
| Allogeneic only | 3 (9.7) | 1 (16.7) | 0 | 1 (16.7) | 0 | 1 (16.7) |
| Autologous and allogeneic | 6 (19.4) | 1 (16.7) | 2 (28.6) | 1 (16.7) | 2 (33.3) | 0 |
| Characteristic | All patients, N = 31, median (range) or n (%) | Avelumab treatment group, median (range) or n (%) | ||||
|---|---|---|---|---|---|---|
| 70 mg Q2W, n = 6 | 350 mg Q2W, n = 7 | 500 mg Q2W, n = 6 | 500 mg Q3W, n = 6 | 10 mg/kg Q2W, n = 6 | ||
| Age, y | 38.0 (22.0-81.0) | 57.0 (31.0-78.0) | 48.0 (30.0-71.0) | 36.5 (22.0-56.0) | 35.0 (25.0-78.0) | 31.5 (25.0-81.0) |
| Age group, y | ||||||
| <65 | 24 (77.4) | 5 (83.3) | 4 (57.1) | 6 (100.0) | 4 (66.7) | 5 (83.3) |
| ≥65 | 7 (22.6) | 1 (16.7) | 3 (42.9) | 0 | 2 (33.3) | 1 (16.7) |
| ECOG performance status | ||||||
| 0 | 14 (45.2) | 2 (33.3) | 4 (57.1) | 3 (50.0) | 3 (50.0) | 2 (33.3) |
| 1 | 17 (54.8) | 4 (66.7) | 3 (42.9) | 3 (50.0) | 3 (50.0) | 4 (66.7) |
| Sex | ||||||
| Male | 24 (77.4) | 3 (50.0) | 6 (85.7) | 5 (83.3) | 5 (83.3) | 5 (83.3) |
| Female | 7 (22.6) | 3 (50.0) | 1 (14.3) | 1 (16.7) | 1 (16.7) | 1 (16.7) |
| Geographic region | ||||||
| North America | 10 (32.2) | 2 (33.3) | 2 (28.6) | 2 (33.3) | 2 (33.3) | 2 (33.3) |
| Western Europe | 21 (67.7) | 4 (66.7) | 5 (71.4) | 4 (66.7) | 4 (66.7) | 4 (66.7) |
| No. of prior anticancer therapy regimens | ||||||
| 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | 2 (6.5) | 0 | 0 | 0 | 1 (16.7) | 1 (16.7) |
| 3 | 9 (29.0) | 2 (33.0) | 2 (28.6) | 1 (16.7) | 2 (33.3) | 2 (33.3) |
| ≥4 | 20 (64.5) | 4 (66.7) | 5 (71.4) | 5 (83.3) | 3 (50.0) | 3 (50.0) |
| Prior treatment with brentuximab vedotin | 31 (100.0) | 6 (100.0) | 7 (100.0) | 6 (100.0) | 6 (100.0) | 6 (100.0) |
| Patients with prior HSCT | 13 (41.9) | 2 (33.3) | 2 (28.6) | 4 (66.7) | 4 (66.7) | 1 (16.7) |
| Type of HSCT | ||||||
| Autologous only | 4 (12.9) | 0 | 0 | 2 (33.3) | 2 (33.3) | 0 |
| Allogeneic only | 3 (9.7) | 1 (16.7) | 0 | 1 (16.7) | 0 | 1 (16.7) |
| Autologous and allogeneic | 6 (19.4) | 1 (16.7) | 2 (28.6) | 1 (16.7) | 2 (33.3) | 0 |
ECOG, Eastern Cooperative Oncology Group; Q2W, every 2 weeks; Q3W, every 3 weeks.
PK profile
The PK analysis population included patients from the lead-in phase. Median PK exposures were dose proportional (Figure 1; supplemental Table 1; supplemental Figure 1). TO was sustained (>90%) throughout the dosing interval, regardless of dose level or schedule.
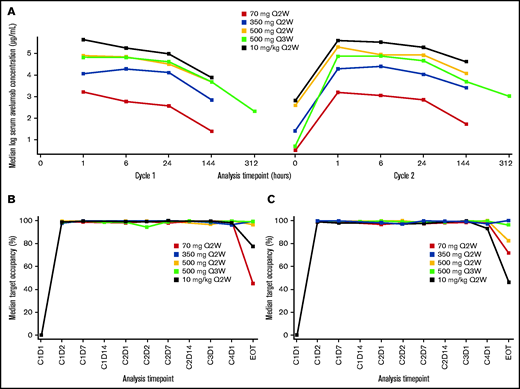
PK and pharmacodynamic analyses. (A) Median serum concentration starting on cycle 1 day 1. (B) Median TO on CD14+ monocytes. (C) Median TO on CD3+ T cells. C, cycle; D, day; EOT, end of treatment; Q2W, every 2 weeks; Q3W, every 3 weeks.
PK and pharmacodynamic analyses. (A) Median serum concentration starting on cycle 1 day 1. (B) Median TO on CD14+ monocytes. (C) Median TO on CD3+ T cells. C, cycle; D, day; EOT, end of treatment; Q2W, every 2 weeks; Q3W, every 3 weeks.
Efficacy
Due to the small number of patients in each treatment/dose group, efficacy results are summarized across all groups. In all patients, the median duration of treatment was 3.9 months (range, 0.5-28.3 months; Table 2). The median duration of treatment in patients with cHL that progressed following auto- or allo-HSCT was 4.3 months (range, 0.9-6.2 months) and 3.0 months (range, 0.5-18.0 months), respectively. The ORR in all randomized patients was 41.9% (95% CI, 24.5% to 60.9%), with a BOR of CR in 6 patients (19.4%) and PR in 7 patients (22.6%). In the 350-mg treatment group, no CRs or PRs were observed. The ORR in patients with cHL that progressed following auto- or allo-HSCT was 25.0% (95% CI, 0.6% to 80.6%), with 1 CR, and 55.6% (95% CI, 21.2% to 86.3%), with 3 CRs, respectively. The median time to response in all patients was 1.5 months (range, 1.4-3.5 months) and ranged from 1.4 months (range, 1.4-2.8 months) in the 10-mg/kg every-2-weeks group to 2.6 months (range, 1.7-3.5 months) in the 70-mg every-2-weeks group (Figure 2). The median duration of response in all patients and in those who progressed following allo-HSCT was 6.9 months (95% CI, 1.4 months to not estimable) and 6.9 months (95% CI, 1.8-12.4 months), respectively (Table 2; Figure 2). Changes in tumor burden are also shown in Figure 2. As of the data cutoff (11 April 2019), the median follow-up for PFS in all patients was 5.6 months (95% CI, 0-11.1 months); median PFS was 5.7 months (95% CI, 2.6-10.8 months) and the 1-year PFS rate was 18.2% (95% CI, 3.3% to 42.5%).
Duration of treatment and antitumor activity
| All patients, N = 31 | Avelumab treatment group | Post– auto-HSCT, n = 4 | Post– allo-HSCT, n = 9* | |||||
|---|---|---|---|---|---|---|---|---|
| 70 mg Q2W, n = 6 | 350 mg Q2W, n = 7 | 500 mg Q2W, n = 6 | 500 mg Q3W, n = 6 | 10 mg/kg Q2W, n = 6 | ||||
| Duration of treatment, median (range), mo | 3.9 (0.5-28.3) | 2.3 (0.5-10.8) | 4.6 (2.1-28.3) | 3.7 (0.9-18.0) | 5.9 (0.7-11.8) | 3.7 (1.8-8.8) | 4.3 (0.9-6.2) | 3.0 (0.5-18.0) |
| BOR, n (%) | ||||||||
| CR | 6 (19.4) | 1 (16.7) | 0 | 0 | 4 (66.7) | 1 (16.7) | 1 (25.0) | 3 (33.3) |
| PR | 7 (22.6) | 1 (16.7) | 0 | 2 (33.3) | 1 (16.7) | 3 (50.0) | 0 | 2 (22.2) |
| SD | 8 (25.8) | 1 (16.7) | 4 (57.1) | 2 (33.3) | 1 (16.7) | 0 | 2 (50.0) | 2 (22.2) |
| PD | 6 (19.4) | 2 (33.3) | 1 (14.3) | 1 (16.7) | 0 | 2 (33.3) | 0 | 1 (11.1) |
| Not evaluable | 4 (12.9) | 1 (16.7)† | 2 (28.6)‡,§ | 1 (16.7)† | 0 | 0 | 1 (25.0)† | 1 (11.1)† |
| ORR (95% CI), % | 41.9 (24.5-60.9) | 33.3 (4.3-77.7) | 0 (0-41.0) | 33.3 (4.3-77.7) | 83.3 (35.9-99.6) | 66.7 (22.3-95.7) | 25.0 (0.6-80.6) | 55.6 (21.2-86.3) |
| Time to response, median (range), mo | 1.5 (1.4-3.5) | 2.6 (1.7-3.5) | — | 1.5 (1.4-1.5) | 1.5 (1.4-1.6) | 1.4 (1.4-2.8) | 1.4 (1.4-1.4) | 1.6 (1.4-3.5) |
| Duration of response, median (95% CI), mo | 6.9 (1.4-NE) | NE (NE-NE) | — | 6.9 (1.4-12.4) | 6.9 (4.3-NE) | 9.4 (1.8-9.4) | NE (NE-NE) | 6.9 (1.8-12.4) |
| All patients, N = 31 | Avelumab treatment group | Post– auto-HSCT, n = 4 | Post– allo-HSCT, n = 9* | |||||
|---|---|---|---|---|---|---|---|---|
| 70 mg Q2W, n = 6 | 350 mg Q2W, n = 7 | 500 mg Q2W, n = 6 | 500 mg Q3W, n = 6 | 10 mg/kg Q2W, n = 6 | ||||
| Duration of treatment, median (range), mo | 3.9 (0.5-28.3) | 2.3 (0.5-10.8) | 4.6 (2.1-28.3) | 3.7 (0.9-18.0) | 5.9 (0.7-11.8) | 3.7 (1.8-8.8) | 4.3 (0.9-6.2) | 3.0 (0.5-18.0) |
| BOR, n (%) | ||||||||
| CR | 6 (19.4) | 1 (16.7) | 0 | 0 | 4 (66.7) | 1 (16.7) | 1 (25.0) | 3 (33.3) |
| PR | 7 (22.6) | 1 (16.7) | 0 | 2 (33.3) | 1 (16.7) | 3 (50.0) | 0 | 2 (22.2) |
| SD | 8 (25.8) | 1 (16.7) | 4 (57.1) | 2 (33.3) | 1 (16.7) | 0 | 2 (50.0) | 2 (22.2) |
| PD | 6 (19.4) | 2 (33.3) | 1 (14.3) | 1 (16.7) | 0 | 2 (33.3) | 0 | 1 (11.1) |
| Not evaluable | 4 (12.9) | 1 (16.7)† | 2 (28.6)‡,§ | 1 (16.7)† | 0 | 0 | 1 (25.0)† | 1 (11.1)† |
| ORR (95% CI), % | 41.9 (24.5-60.9) | 33.3 (4.3-77.7) | 0 (0-41.0) | 33.3 (4.3-77.7) | 83.3 (35.9-99.6) | 66.7 (22.3-95.7) | 25.0 (0.6-80.6) | 55.6 (21.2-86.3) |
| Time to response, median (range), mo | 1.5 (1.4-3.5) | 2.6 (1.7-3.5) | — | 1.5 (1.4-1.5) | 1.5 (1.4-1.6) | 1.4 (1.4-2.8) | 1.4 (1.4-1.4) | 1.6 (1.4-3.5) |
| Duration of response, median (95% CI), mo | 6.9 (1.4-NE) | NE (NE-NE) | — | 6.9 (1.4-12.4) | 6.9 (4.3-NE) | 9.4 (1.8-9.4) | NE (NE-NE) | 6.9 (1.8-12.4) |
—, not applicable. NE, not estimable. See Table 1 for expansion of other abbreviations.
Patient count includes the 6 patients who received both auto-HSCT and allo-HSCT.
All postbaseline assessments had an overall response of not evaluable.
No adequate baseline assessment (n = 1).
SD too early (<6 weeks after randomization; n = 1).
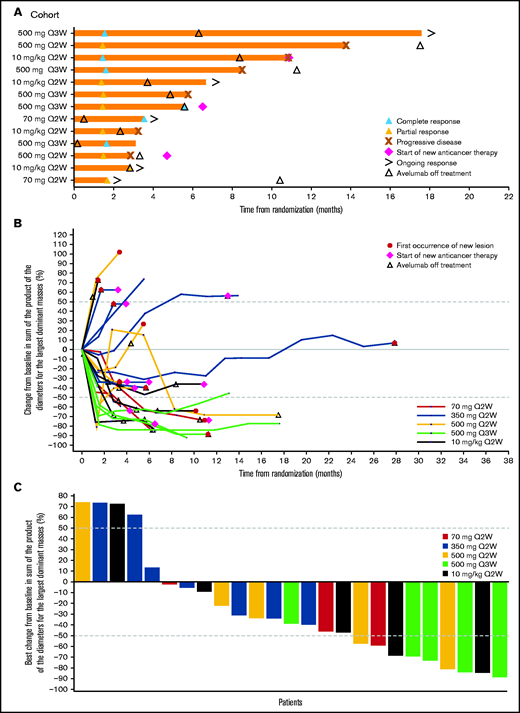
Clinical responses to avelumab. (A) Time to and duration of response among patients with an objective response across treatment groups per investigator assessment. (B) Percentage change in tumor burden per investigator assessment. (C) Best percentage change in tumor burden per investigator assessment. (B-C) Only patients with the largest dominant masses at baseline and ≥1 postbaseline assessment are included. The last measurement prior to the randomization date served as the baseline measurement. Patients missing from this summary were included in the full analysis set but were excluded here due to missing data: no baseline measurements within the 28-day window (n = 2); no appropriate postbaseline measurements (n = 3). Q2W, every 2 weeks; Q3W, every 3 weeks.
Clinical responses to avelumab. (A) Time to and duration of response among patients with an objective response across treatment groups per investigator assessment. (B) Percentage change in tumor burden per investigator assessment. (C) Best percentage change in tumor burden per investigator assessment. (B-C) Only patients with the largest dominant masses at baseline and ≥1 postbaseline assessment are included. The last measurement prior to the randomization date served as the baseline measurement. Patients missing from this summary were included in the full analysis set but were excluded here due to missing data: no baseline measurements within the 28-day window (n = 2); no appropriate postbaseline measurements (n = 3). Q2W, every 2 weeks; Q3W, every 3 weeks.
Safety
At data cutoff, all patients had discontinued avelumab; the most common reasons for treatment discontinuation were progressive disease (n = 15 [48.4%]) and adverse events (AEs) (n = 6 [19.4%]). The incidence of AEs across the 5 treatment groups was similar, and the safety profile is therefore presented for all treated patients together (N = 30). Any-grade treatment-related AEs (TRAEs) occurred in 26 patients (86.7%) (Table 3). The most common TRAEs occurring in >10% of patients were infusion-related reaction (n = 9 [30.0%]), nausea (n = 6 [20.0%]), alanine aminotransferase increased and rash (n = 5 each [16.7%]), and fatigue (n = 4 [13.3%]). Grade ≥3 TRAEs occurred in 13 patients (43.3%). Grade 3/4 TRAEs were mostly laboratory abnormalities, and no single grade 3/4 TRAE was reported in >2 patients. One treatment-related death (pneumonitis) occurred in the avelumab 350-mg every-2-weeks treatment group.
TRAEs in all treated patients
| N = 30, n (%) | |||||
|---|---|---|---|---|---|
| Any grade | Grade 1 | Grade 2 | Grade 3 | Grade 4 | |
| Patients with events | 26 (86.7) | 4 (13.3) | 9 (30.0) | 9 (30.0) | 4 (13.3) |
| Infusion-related reaction* | 9 (30.0) | 1 (3.3) | 6 (20.0) | 2 (6.7) | 0 |
| Nausea | 6 (20.0) | 5 (16.7) | 0 | 1 (3.3) | 0 |
| Alanine aminotransferase increased | 5 (16.7) | 3 (10.0) | 0 | 2 (6.7) | 0 |
| Rash | 5 (16.7) | 4 (13.3) | 1 (3.3) | 0 | 0 |
| Fatigue | 4 (13.3) | 4 (13.3) | 0 | 0 | 0 |
| Aspartate aminotransferase increased | 3 (10.0) | 2 (6.7) | 0 | 1 (3.3) | 0 |
| Back pain | 3 (10.0) | 0 | 2 (6.7) | 1 (3.3) | 0 |
| Blood alkaline phosphatase increased | 3 (10.0) | 0 | 1 (3.3) | 2 (6.7) | 0 |
| Constipation | 3 (10.0) | 3 (10.0) | 0 | 0 | 0 |
| Diarrhea | 3 (10.0) | 3 (10.0) | 0 | 0 | 0 |
| Lipase increased | 3 (10.0) | 0 | 1 (3.3) | 0 | 2 (6.7) |
| Abdominal pain | 2 (6.7) | 2 (6.7) | 0 | 0 | 0 |
| Amylase increased | 2 (6.7) | 0 | 0 | 2 (6.7) | 0 |
| GVHD in liver | 2 (6.7) | 0 | 0 | 2 (6.7) | 0 |
| Biliary sepsis | 1 (3.3) | 0 | 0 | 1 (3.3) | 0 |
| C-reactive protein increased | 1 (3.3) | 0 | 0 | 1 (3.3) | 0 |
| Encephalitis | 1 (3.3) | 0 | 0 | 1 (3.3) | 0 |
| Immune thrombocytopenic purpura | 1 (3.3) | 0 | 0 | 0 | 1 (3.3) |
| Jaundice | 1 (3.3) | 0 | 0 | 1 (3.3) | 0 |
| Orthostatic hypotension | 1 (3.3) | 0 | 0 | 1 (3.3) | 0 |
| Pneumonitis | 1 (3.3) | 0 | 0 | 1 (3.3) | 0 |
| Thrombocytopenia | 1 (3.3) | 0 | 0 | 0 | 1 (3.3) |
| Transaminases increased | 1 (3.3) | 0 | 0 | 1 (3.3) | 0 |
| N = 30, n (%) | |||||
|---|---|---|---|---|---|
| Any grade | Grade 1 | Grade 2 | Grade 3 | Grade 4 | |
| Patients with events | 26 (86.7) | 4 (13.3) | 9 (30.0) | 9 (30.0) | 4 (13.3) |
| Infusion-related reaction* | 9 (30.0) | 1 (3.3) | 6 (20.0) | 2 (6.7) | 0 |
| Nausea | 6 (20.0) | 5 (16.7) | 0 | 1 (3.3) | 0 |
| Alanine aminotransferase increased | 5 (16.7) | 3 (10.0) | 0 | 2 (6.7) | 0 |
| Rash | 5 (16.7) | 4 (13.3) | 1 (3.3) | 0 | 0 |
| Fatigue | 4 (13.3) | 4 (13.3) | 0 | 0 | 0 |
| Aspartate aminotransferase increased | 3 (10.0) | 2 (6.7) | 0 | 1 (3.3) | 0 |
| Back pain | 3 (10.0) | 0 | 2 (6.7) | 1 (3.3) | 0 |
| Blood alkaline phosphatase increased | 3 (10.0) | 0 | 1 (3.3) | 2 (6.7) | 0 |
| Constipation | 3 (10.0) | 3 (10.0) | 0 | 0 | 0 |
| Diarrhea | 3 (10.0) | 3 (10.0) | 0 | 0 | 0 |
| Lipase increased | 3 (10.0) | 0 | 1 (3.3) | 0 | 2 (6.7) |
| Abdominal pain | 2 (6.7) | 2 (6.7) | 0 | 0 | 0 |
| Amylase increased | 2 (6.7) | 0 | 0 | 2 (6.7) | 0 |
| GVHD in liver | 2 (6.7) | 0 | 0 | 2 (6.7) | 0 |
| Biliary sepsis | 1 (3.3) | 0 | 0 | 1 (3.3) | 0 |
| C-reactive protein increased | 1 (3.3) | 0 | 0 | 1 (3.3) | 0 |
| Encephalitis | 1 (3.3) | 0 | 0 | 1 (3.3) | 0 |
| Immune thrombocytopenic purpura | 1 (3.3) | 0 | 0 | 0 | 1 (3.3) |
| Jaundice | 1 (3.3) | 0 | 0 | 1 (3.3) | 0 |
| Orthostatic hypotension | 1 (3.3) | 0 | 0 | 1 (3.3) | 0 |
| Pneumonitis | 1 (3.3) | 0 | 0 | 1 (3.3) | 0 |
| Thrombocytopenia | 1 (3.3) | 0 | 0 | 0 | 1 (3.3) |
| Transaminases increased | 1 (3.3) | 0 | 0 | 1 (3.3) | 0 |
Events of any grade in ≥2 patients and all grade ≥3 events are shown.
Infusion-related reaction is a composite term that includes infusion-related reaction, back pain, chills, and pyrexia.
The overall safety profile of avelumab in the 8 patients with prior allo-HSCT who received treatment was generally consistent with that in patients who had not received prior allo-HSCT, with a few notable exceptions. One patient in the 70-mg every-2-weeks group who had prior GVHD in the liver received 2 doses of avelumab and developed grade 3 GVHD in the liver 22 days after the start of treatment; avelumab was discontinued. The patient received immunosuppressive therapy with methylprednisolone, basiliximab, mycophenolate mofetil, sirolimus, and infliximab. At data cutoff, GVHD in the liver had not resolved. In addition, 1 patient in the 500-mg every-3-weeks group without prior GVHD received 1 dose of avelumab and developed grade 3 GVHD in the liver 21 days after the start of treatment; avelumab was discontinued. The patient received methylprednisolone and rituximab, and the event of GVHD in the liver resolved. Approximately 8 months after starting avelumab, the patient developed grade 3 chronic GVHD in the skin and received prednisolone, cyclosporine, imatinib, and extracorporeal photopheresis. The patient eventually died of pseudomonal sepsis. Both patients achieved a CR.
Any-grade immune-related AEs (irAEs; identified according to a predefined case definition) were observed in 9 patients (30.0%) (supplemental Table 2). The most frequent irAEs were rash (n = 2 [6.7%]) and GVHD in the liver occurring in the same 2 patients. Grade ≥3 irAEs occurred in 5 patients (16.7%). One patient died of immune-related pneumonitis 56 days after the last dose of avelumab. Other grade ≥3 irAEs included transaminases increased and immune thrombocytopenic purpura (n = 1 each [3.3%]).
Biomarker analyses
Four patients had sufficient tumor tissue from baseline tumor biopsies for assessment of PD-L1/PD-L2 by immunohistochemistry (IHC) and FISH. All samples were positive for PD-L1 by IHC; however, no effective PD-L2 IHC reagent could be found. Of 4 FISH-evaluable samples, 2 showed PD-L1/PD-L2 copy-number alterations (supplemental Figure 2) and 1 had polysomy; the other had amplification. Both patients had a PR as a BOR. The remaining 2 patients had a BOR of CR and PD, respectively.
Expansion phase
Three patients were enrolled in the expansion phase (avelumab 70 mg every 2 weeks, with possible escalation to 500 mg every 2 weeks). All patients had received prior auto- and allo-HSCT. The first patient was a 40-year-old woman who received 24 cycles of avelumab starting with 70 mg, with escalation to 500 mg at cycle 4 (treatment duration, 46.29 weeks). Her BOR was stable disease, and she experienced 1 TRAE (pyrexia, grade 1). The second patient was a 24-year-old man who received 1 cycle of avelumab 70 mg (treatment duration, 0.14 weeks). BOR was not evaluable due to lack of adequate baseline assessment, and he did not experience any TRAEs. The third patient was a 24-year-old woman who received 13 cycles of avelumab starting with 70 mg, with escalation to 500 mg at cycle 8 (treatment duration, 24.14 weeks). Her BOR was a PR, and she experienced 2 grade 2 TRAEs (dyspnea and erythema).
Discussion
This trial investigated multiple dosing schedules of avelumab in patients with R/R cHL to gain insight into the dosing regimen(s) that would provide optimal TO over the entire dosing interval and to help characterize the dose exposure-response profile of avelumab. In this study, the PK of avelumab was dose proportional across all dose cohorts, with geometric mean Ctrough concentrations above 1 μg/mL and similar PK characteristics for dosing intervals of every 2 weeks and every 3 weeks. Full TO (>90%) was sustained throughout the dosing interval. In alignment with the TO data, objective responses were observed across the range of avelumab exposure levels. Given the dose proportionality of PK and the maintenance of TO, it is not clear what parameters might have accounted for the lack of responses in the 350-mg every-2-weeks group. It should be noted that TO was measured on circulating immune cells as a surrogate for direct measurement on tumor cells, on which PD-L1 expression levels may be higher, so it is possible that factors such as differences in tumor PD-L1 expression levels, tumor burden, and treatment history across the small patient groups could have contributed to differential degrees of antitumor activity. Moreover, there is potential for alteration in PD-L1 expression as a consequence of PD-1/PD-L1 blockade, which might complicate assessment of the fraction of free PD-L1.27 In addition, the sample size in each group was too small to determine whether 1 regimen was significantly more clinically active than another.
For all treatment groups combined, the ORR of 41.9% in these heavily pretreated patients with R/R cHL is lower than the ORR range observed with nivolumab (from 69% to 87%)8,28 or pembrolizumab (from 58% to 72%)12,29 in similar patient populations with a median of ≥4 prior lines of systemic therapy. The CR rate of 19.4% with avelumab in all treatment groups combined in this study was similar to that seen with nivolumab (16% to 17%)8,28 and pembrolizumab (19% to 27.6%).12,29 The PR rate of 22.6% in this study was lower than in comparable studies (ranging from 39% to 70%). The median duration of response (6.9 months) and median PFS (5.7 months) in this study were shorter than those observed in comparable studies (∼16.5 months and 11.4 to 14.7 months, respectively).8,12,28,29 This may be due to the small sample size in our study, differences in characteristics of the treated patients (eg, tumor burden, inclusion of patients who received prior allo-HSCT), or the differing doses and dosing schedules used in this study. In contrast to nivolumab and pembrolizumab, which target PD-1 and inhibit its interaction with PD-L1 and PD-L2, avelumab binds PD-L1 and inhibits its interaction with PD-1 but leaves the PD-1/PD-L2 interaction intact. Amplification of chromosome 9p24.1 is frequently observed in cHL,6,7 leading to enhanced expression of PD-L1 and PD-L2. Avelumab showed clinical activity in patients with heavily pretreated cHL, suggesting that PD-L1 blockade is sufficient to produce clinical responses. PD-L2 blockade may not be necessary to achieve the therapeutic effect observed with PD-1 inhibitors in some patients. However, a role for PD-L2 blockade in dampening the therapeutic effect of PD-L1/PD-1 blockade in some patients cannot be ruled out and could provide an alternative explanation for the lower ORR, duration of response, and PFS observed in this study compared with studies reported for PD-1–blocking agents.
The AEs observed in this study were generally consistent with the known safety profile of avelumab.30 One key question that was addressed in this study was the safety of treatment of patients in a post–allo-HSCT setting with a specific PD-1/PD-L1 inhibitor. Similar to what has been observed in prior retrospective studies, grade 3 acute liver GVHD occurred in 2 of 9 patients in this study who had previously received allo-HSCT; however, immunosuppressive therapy resulted in complete resolution of these events in 1 of 2 patients. Although our prospective clinical trial population is likely different (exclusions for prior severe acute GVHD, persistent chronic GVHD, or recent immunosuppression) than the patients reported in retrospective studies, our results compare favorably with those of other studies that have reported GVHD rates of 30% to 55% and mortality rates of 10% to 26% due to GVHD in patients who received PD-1 blockade after allo-HSCT.11,12 Notably, both patients who developed GVHD in our study achieved a CR, which suggests that avelumab may have stimulated a graft-versus-tumor response in these patients. Whether the higher ORR of 55.6% observed in the 9 patients with prior allo-HSCT who received avelumab is reflective of better efficacy than in patients without a prior HSCT due to stimulation of a graft-versus-tumor response is unclear; however, PD-L1 blockade can result in antitumor responses in this patient population similar to what has been observed in other studies.11,12 Due to decreased use of allo-HSCT in patients with R/R cHL, the post–allo-HSCT setting could not be further explored in this study, and the expansion phase was terminated after the enrollment of 3 patients.
In conclusion, avelumab showed antitumor activity and tolerable safety in heavily pretreated patients with cHL. The use of PD-1 blockade in the treatment of patients with cHL has become a standard of care, but anti–PD-1 monotherapy is associated with a durable CR in a minority of patients. Therefore, improving the depth and durability of responses to checkpoint blockade therapy in R/R cHL remains an unmet need.
Acknowledgments
The authors thank the patients and their families and the investigators, coinvestigators, and study teams at each of the participating centers. The authors also thank Eric Negre for substantial contributions to this work, Adrian Woolfson and Giovanna Andreola for their role in the design and conduct of the study, and Allison Forgie for contributions to the biomarker analyses.
W.T. acknowledges support and funding from the National Institute for Health Research University College Hospitals Biomedical Research Centre.
This trial was sponsored by Pfizer Inc, as part of an alliance between Pfizer and Merck KGaA, Darmstadt, Germany. Medical writing support was provided by Shaun Rosebeck of ClinicalThinking, funded by Pfizer and Merck KGaA. G.P.C. acknowledges support from the NIHR Oxford Biomedical Research Centre and the Cancer Research UK Experimental Cancer Medicine Centre. A.F.H. was supported by the Lymphoma Research Foundation’s Larry and Denise Mason Clinical Investigator Career Development Award and a Toni and Emmet Stephenson Leukemia & Lymphoma Society Scholar in Clinical Research Award.
Authorship
Contribution: A.F.H., C.B., J.R., F.M., W.T., A.S., P.L.Z., D.L., and G.P.C. collected, assembled, analyzed, and interpreted the data; C.F. and A.T. conceived and designed the study and analyzed and interpreted the data; S.B. and B.H. conceived and designed the study and collected, assembled, analyzed, and interpreted the data; and all authors were involved in writing the article and approved the final version.
Conflict-of-interest disclosure: A.F.H. reports a consultancy/advisory role for Bristol Myers Squibb, Merck & Co, Karyopharm, and Seattle Genetics; and research funding from Bristol Myers Squibb, Genentech, Merck & Co, Pharmacyclics LLC, an AbbVie Company, Kite Pharma, Seattle Genetics, Immune Design, and AstraZeneca. J.R. reports a consultancy/advisory role for Takeda, Bristol Myers Squibb, ADC Therapeutics, and Novartis; owns stock in AstraZeneca and ADC Therapeutics; reports honoraria from Takeda, ADC Therapeutics, and Bristol Myers Squibb; provides speaker/expert testimony for Takeda and ADC Therapeutics; and reports research funding from Takeda. F.M. has received honoraria fees from Takeda and Roche. W.T. has received honoraria and consultancy fees from Roche and Gilead. A.S. reports a consultancy/advisory role for Bristol Myers Squibb, Servier, Gilead, Pfizer, Eisai, Bayer, Merck Sharp & Dohme (MSD), Sanofi, and ArQule; and speakers’ bureau work for Takeda, Roche, AbbVie, Amgen, Celgene, AstraZeneca, ArQule, Eli Lilly, Sandoz, Novartis, Bristol Myers Squibb, Servier, Gilead, Pfizer, Eisai, Bayer, and MSD. P.L.Z. reports a consultancy/advisory role for Verastem, Gilead, Janssen-Cilag, Bristol Myers Squibb, Servier, Sandoz, MSD, Immune Design, Celgene, Portola, Roche, EUSA Pharma, Kyowa Kirin, and Sanofi; and speakers’ bureau work for Verastem, Gilead, Janssen-Cilag, Bristol Myers Squibb, Servier, MSD, Immune Design, Celgene, Portola, Roche, EUSA Pharma, and Kyowa Kirin. C.F., S.B., B.H., and A.T. are employees of Pfizer and report stock ownership in Pfizer. G.P.C. has received honoraria for advisory work and research funding from Pfizer; honoraria for advisory and speakers’ bureau work from Roche, Takeda, Gilead, Bristol Myers Squibb, MSD, Celleron, ADC Therapeutics, and BeiGene; and research funding from Bristol Myers Squibb, MSD, Celleron, Amgen, and Celgene. The remaining authors declare no competing financial interests.
Correspondence: Alex F. Herrera, City of Hope Medical Center, 1500 East Duarte Rd, Duarte, CA 91010; e-mail: aherrera@coh.org.

