

Key Points
del17p is less frequent in AAs than in White patients with MM.
With equal access to care, del17p carries poor prognosis across race; however, AAs with non-del17p MM still have superior OS.
Abstract
Multiple myeloma (MM) is a heterogeneous disease that has an increased incidence in African Americans (AAs). We previously observed that, with equal access to health care, younger AA patients (age < 65 years) have superior overall survival (OS) compared with younger White patients. Because MM prognosis is influenced by 17p deletion (del17p), we investigated racial differences in its occurrence and impact in a large cohort of MM patients from the Veterans Affairs (VA) system. Among 2243 VA patients with MM for whom del17p data were available, del17p was present in 8.83% of all patients, with a significantly lower prevalence in AAs (5.56%) compared with Whites (10.52%; P < .001). The difference was even more pronounced among younger AAs (<65 years) vs younger Whites (4.34% vs 9.8%, respectively; P = .004). However, we did not observe any significant difference in survival between AA and White patients with del17p, regardless of age category, suggesting that del17p carries a poor prognosis across race and age. Interestingly, among patients without del17p, we still noted a significantly superior OS in younger AAs compared with younger Whites (7.75 vs 5.10 years; P = .042). Our study shows a lower incidence of del17p in AAs but suggests that the survival advantage for younger AAs is primarily due to factors other than del17p.
Introduction
Multiple myeloma (MM)1 is the second most common hematologic malignancy in the United States and the most common hematologic malignancy in African Americans (AAs), constituting one of the cancers with the greatest racial disparity.2,3 AAs have more than twice the incidence of MM and an earlier age of onset (by 5-10 years) compared with White patients.4 This difference is even greater in those younger than 50 years of age, largely because of the greater prevalence of the myeloma precursor state, monoclonal gammopathy of undetermined significance (MGUS), in AAs (0.88% vs 0.22%; P = .001), with up to 10 years earlier age of MGUS onset in AAs compared with Whites.5 Studies suggest that racial disparities in MGUS may be related to genetic differences.1,6 Although 17p deletion (del17p) is present in only ∼10% of newly diagnosed MM patients, its presence is an independent poor-prognostic factor that is associated with shortened OS (hazard ratio, 2.03) and progression-free survival (hazard ratio,1.82).7-11 Here, we investigated the prevalence of del17p and its impact on outcome in AA and White patients with MM using the large Veterans Affairs (VA) system database to understand the racial prevalence and any racial difference in its clinical significance.
Methods
Patients diagnosed with MM in the VA’s nationwide database of electronic health records between 1999 and 2019 were identified using the VA Corporate Data Warehouse, as described in our previous work.12,13 Patient demographics, as well as drugs and therapies used, were identified through structured data in the Corporate Data Warehouse. We identified del17p using pattern matching and natural language processing techniques from pathology or laboratory test reports from bone marrow performed up to 90 days after diagnosis. This study was approved by the VA Boston Healthcare System Research and Development Committee and was conducted in accordance with the Declaration of Helsinki.
Results and discussion
Of 15717 patients diagnosed with MM, 2243 were evaluated for del17p by conventional cytogenetics and/or fluorescence in situ hybridization (FISH). Based on self-reported race information, 33% (737) of patients were AA, 58% (1312) were White, and 9% (194) were of other or unknown race; based on self-reported ethnicity, 9% (204) of patients were Hispanic or Latino. AAs had a greater proportion of younger (<65 years) patients compared with Whites (50.01% vs 34.22%; P < .001), consistent with our prior reports. As in our prior report, we did not observe racial disparity with regard to the overall use of novel agents at induction or stem cell transplant.14 Reflecting veteran demographics, 97.5% of patients were male, and 70.3% were urban. Compared with the entire cohort, 23% of AA patients and 15% of White patients were evaluated by FISH; in general, patients tested using FISH were diagnosed more recently (median year of diagnosis, 2013 vs 2007; P < .001).
Overall, among those tested, del17p was reported in 8.83% (198/2243) of all patients, but the prevalence was significantly lower in AAs compared with Whites (5.56% vs 10.52%; P < .001), with a prevalence of 3.92% in Hispanics. Previous studies noted a lower frequency of del17p in AAs both at the MGUS stage6 and at progression to MM1,14,15 ; however, those findings either lacked significance or lacked correlation with clinical characteristics and survival. Furthermore, our analysis revealed that the discrepancy in the prevalence of del17p was more pronounced among younger (<65 years) AAs vs younger Whites (younger: 4.34% vs 9.8%, respectively; P = .004; older: 6.79% vs 10.89%, respectively; P = .0338). The rates of del17p across different International Staging System (ISS) stages were not significantly different (P = .215), and ISS stage did not differ across race (P = .068).
As expected, del17p was associated with shortened survival. In the entire cohort, patients with del17p (n = 198) had shorter median overall survival compared with those without del17p (n = 2045; 2.26 vs 4.49 years, respectively; P < .0001; Figure 1A). These differences were also observed within race (1.74 vs 5.64 years in AAs; P < .0001 and 2.34 vs 4.37 years in Whites, P < .0001) and age groups (2.67 vs 5.75 years in younger patients; P < .0001 and 2.12 vs 3.64 years in older patients; P < .0001), but not in Hispanic patients, although the sample size was small (2.92 vs 3.51 years; P = .26).
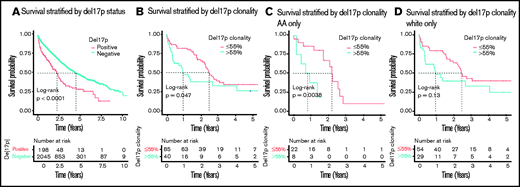
OS, stratified by del17p status and clonality. (A) OS in the entire cohort of patients tested for del17p. (B) OS in the group of all del17p+ patients with a reported clonality. (C-D) OS by del17p clonality stratified by race.
OS, stratified by del17p status and clonality. (A) OS in the entire cohort of patients tested for del17p. (B) OS in the group of all del17p+ patients with a reported clonality. (C-D) OS by del17p clonality stratified by race.
Although our data did not allow for evaluation for biallelic inactivation of TP53, which can also have a significant impact on patient outcome,16 we were able to evaluate the impact of del17p clonality. Among 198 patients with del17p, 125 (63%) had the percentage of cells carrying del17p reported. Eighty-five patients (68%) had a clonal cell fraction ≤ 55%, whereas 40 patients (32%) had a clonal cell fraction > 55%. The median clonal fraction was similar across race (median, 25%; interquartile range, 11-65 in AAs vs 39%; interquartile range, 16-72 in Whites; P = .24). We observed that higher clonality (>55%) was associated with lower median survival compared with lower clonality (5-55%) (0.97 vs 2.45 years; P = .047) (Figure 1B). This replicates recent findings that del17p clonality > 55% is associated with worse prognosis,17 and we observed that the result was consistent across race (0.83 vs 2.23 years in AAs; P = .0038 and 0.97 vs 2.45 years in Whites; P = .13; Figure 1C-D).
In our recent study of outcomes in all MM patients treated at the VA from 1999 to 2017, we found that younger AAs (<65 years) have superior OS compared to younger White patients, whereas older AAs (≥65 years) have OS that is similar to older White patients12 (Figure 2A,D). Here, we sought to determine whether the improved OS observed in AAs is also reflected in those with del17p. We did not observe any significant difference in survival between AA and White patients with del17p, regardless of age category (2.26 vs 3.07 years in younger patients; P = .57 and 1.71 vs 2.20 years in older patients; P = .52; Figure 2B,E), suggesting that del17p carries a poor prognosis across race and age. However, among patients without del17p, we still noted a significantly superior OS in younger AAs compared with younger Whites (7.75 vs 5.10 years; P = .042), suggesting that the survival advantage for younger AAs is primarily due to factors other than del17p (Figure 2C). In older patients without del17p, there was still no difference in survival between AA and White patients with MM (3.71 vs 3.66 years; P = .49) (Figure 2F).
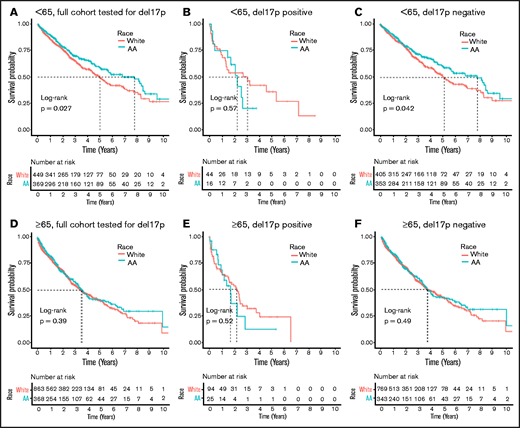
OS stratified by race. The upper row (A-C) shows OS in younger patients (<65 years old at diagnosis with MM), with and without del17p. The lower row (D-F) shows OS in older patients (≥65 years old at diagnosis with MM). The first column (A,D) shows OS in the full cohort of patients tested for del17p,and the other columns show OS in patients who are positive (B,E) and negative (C,F) for del17p.
OS stratified by race. The upper row (A-C) shows OS in younger patients (<65 years old at diagnosis with MM), with and without del17p. The lower row (D-F) shows OS in older patients (≥65 years old at diagnosis with MM). The first column (A,D) shows OS in the full cohort of patients tested for del17p,and the other columns show OS in patients who are positive (B,E) and negative (C,F) for del17p.
To further understand the advantage in survival for younger AA compared with younger White MM patients without del17p, we compared ISS stage, Eastern Cooperative Oncology Group status, therapy usage (bortezomib, lenalidomide, and thalidomide), and stem cell transplant utilization in younger AA and younger White patients. We did not observe any significant difference in these categories other than a small difference in the utilization of bortezomib in younger AA patients compared with younger White patients at induction (63.1% vs 58.3%; P = .029).
To the best of our knowledge, this is the largest cohort tested for the prevalence and impact of del17p on outcome in AA patients with MM. Our study identified a significantly lower prevalence of del17p in AA patients compared with White patients. These results are consistent with previous studies reporting a lower prevalence of del17p (6.7% vs 13.7%) in AAs compared with Whites.7
Moreover, our study confirmed that del17p positivity and higher (>55%) clonality confers a worse prognosis, irrespective of race or age, among patients with equal access to care. The lower prevalence of del17p in AAs only partly explains the superior outcome reported earlier in this racial group. Younger AA patients without del17p MM continue to have superior survival. Thus, our findings suggest possible racial differences in disease biology other than del17p that may contribute to a more indolent course and increased sensitivity to therapy. Studies assessing the role of other cytogenetic factors such as t(4;14), del(1p), or 1q gain, as well as mutational and transcriptomic profiles, may provide additional clues in regard to racial differences. However, these data are not readily available in our system at present. Because del17p has an independent risk association, we believe that the data reported here represents an important contribution. Our study also highlights the need for inclusion of AA patients in clinical studies because observed differences in OS may represent heterogeneity in biology, as well as response to therapy and, potentially, toxicity.
Acknowledgments
This work was supported by the VA Office of Research and Development, Cooperative Studies Program; VA Merit Review Award 1I01BX001584 (N.C.M.); and National Institutes of Health, National Cancer Institute grants P01-155258-07 and P50-100707 (N.C.M.).
The views expressed are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs or the US government.
Authorship
Contribution: N.R.F., D.C., and N.C.M. conceived and designed the study; N.R.F. and A.M. prepared and analyzed the data; S.V.Y., H.Y., N.V.D., M.T.B., and R.E.S. provided scientific input; N.R.F., D.C., A.M., R.E.S. and N.C.M. wrote the manuscript; and all authors edited and critically reviewed the final version of the manuscript.
Conflict-of-interest disclosure: S.V.Y. receives research funding from Takeda and Celgene. M.T.B. receives research funding from Novartis. N.C.M. is consultant for BMS, Janssen Pharmaceuticals, AbbVie, Takeda, Amgen, Novartis, and OncoPep and is a shareholder in OncoPep. The remaining authors declare no competing financial interests.
Correspondence: Nikhil C. Munshi, Jerome Lipper Multiple Myeloma Center, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA 02115; e-mail: nikhil_munshi@dfci.harvard.edu.

