

Abstract
Advances in understanding the ways in which the immune system fails to control tumor growth or prevent autoimmunity have led to the development of powerful therapeutic strategies to treat these diseases. In contrast to conventional therapies that have a broadly suppressive effect, immunotherapies are more akin to targeted therapies because they are mechanistically driven and are typically developed with the goal of “drugging” a specific underlying pathway or phenotype. This means that their effects and toxicities are, at least in theory, more straightforward to anticipate. The development of functionalized antibodies, genetically engineered T cells, and immune checkpoint inhibitors continues to accelerate, illuminating new biology and bringing new treatment to patients. In the following sections, we provide an overview of immunotherapeutic concepts, highlight recent advances in the field of immunotherapies, and discuss controversies and future directions, particularly as these pertain to hematologic oncology or blood-related diseases. We conclude by illustrating how original research published in this journal fits into and contributes to the overall framework of advances in immunotherapy.
Monoclonal antibodies
Antibody-based immunotherapy is among the most successful and well-validated treatment strategies in cancer. The mechanism of action is either direct cell killing (for example, induction of apoptosis), mimicry of basic biologic functions (recruitment of Fc-bearing effector cells that act by antibody-dependent cellular cytotoxicity [ADCC]; antibody-dependent cell phagocytosis [ADCP]; or activation of complement-dependent cytotoxicity [CDC]), or T-cell immunomodulation through blockade or activation of inhibitory or activating immune receptors. In hematologic malignancies, monoclonal antibody (mAb) targets are differentiation antigens that are expressed at distinct maturation steps of a given lineage (lineage-specific antigens [LSA]), or immunoreceptors,1 as shown in Table 1.
FDA-approved therapeutic mAbs and conjugates for cancer therapy
| Target | mAb | MOA | Indication | Reference |
|---|---|---|---|---|
| CCR4 | Mogamulizumab | ADCC | Mycosis fungoides, Sézary syndrome (approved in 2018) | 2 |
| CD19 | Tafasitamab | ADCC/ADCP | DLBCL (approved 2020) | 3 |
| CD20 | Rituximab | CDC/ADCC/PCD | B-NHL, DLBCL, CLL, FL (approved in 1997, 2006, 2010, 2011) | 4–7 |
| CD20 | Ofatumumab | CDC/ADCC/PCD | CLL (approved in 2009) | 8 |
| CD20 | Obinutuzumab | CDC/ADCC/PCD | CLL, FL (approved in 2013, 2016) | 9, 10 |
| CD38 | Daratumumab | ADCC / CDC / ADCP Blockade of CD38 | MM (approved in 2016) | 11 |
| CD38 | Isatuximab | CDC/ADCP/ADCC | MM (approved in 2020) | 12 |
| CD52 | Alemtuzumab | ADCC/CDC/ADCP | B-CLL (approved in 2001) | 13, 14 |
| SLAMF7 | Elotuzumab | ADCC | MM (approved in 2015) | 15 |
| BCMA | Belantamab mafodotin-blmf | ADC | MM (approved in 2020) | 16 |
| CD20 | Ibritumomab tiuxetan | ADC | B-NHL, FL (approved in 2002) | 17 |
| CD22 | Inotuzumab ozogamicin | ADC | ALL (approved in 2017) | 18 |
| CD22 | Moxetumomab pasudotox | ADC | HCL (approved in 2018) | 19 |
| CD30 | Brentuximab vedotin | ADC | HL, ALCL (approved in 2011, 2018) | 20, 21 |
| CD33 | Gemtuzumab ozogamicin | ADC | AML (approved in 2017, 2020) | 22, 23 |
| CD79b | Polatuzumab vedotin | ADC | DLBCL (approved in 2019) | 24 |
| Target | mAb | MOA | Indication | Reference |
|---|---|---|---|---|
| CCR4 | Mogamulizumab | ADCC | Mycosis fungoides, Sézary syndrome (approved in 2018) | 2 |
| CD19 | Tafasitamab | ADCC/ADCP | DLBCL (approved 2020) | 3 |
| CD20 | Rituximab | CDC/ADCC/PCD | B-NHL, DLBCL, CLL, FL (approved in 1997, 2006, 2010, 2011) | 4–7 |
| CD20 | Ofatumumab | CDC/ADCC/PCD | CLL (approved in 2009) | 8 |
| CD20 | Obinutuzumab | CDC/ADCC/PCD | CLL, FL (approved in 2013, 2016) | 9, 10 |
| CD38 | Daratumumab | ADCC / CDC / ADCP Blockade of CD38 | MM (approved in 2016) | 11 |
| CD38 | Isatuximab | CDC/ADCP/ADCC | MM (approved in 2020) | 12 |
| CD52 | Alemtuzumab | ADCC/CDC/ADCP | B-CLL (approved in 2001) | 13, 14 |
| SLAMF7 | Elotuzumab | ADCC | MM (approved in 2015) | 15 |
| BCMA | Belantamab mafodotin-blmf | ADC | MM (approved in 2020) | 16 |
| CD20 | Ibritumomab tiuxetan | ADC | B-NHL, FL (approved in 2002) | 17 |
| CD22 | Inotuzumab ozogamicin | ADC | ALL (approved in 2017) | 18 |
| CD22 | Moxetumomab pasudotox | ADC | HCL (approved in 2018) | 19 |
| CD30 | Brentuximab vedotin | ADC | HL, ALCL (approved in 2011, 2018) | 20, 21 |
| CD33 | Gemtuzumab ozogamicin | ADC | AML (approved in 2017, 2020) | 22, 23 |
| CD79b | Polatuzumab vedotin | ADC | DLBCL (approved in 2019) | 24 |
ADC, antibody-drug-conjugate; ALCL, anaplastic large cell lymphoma; B-NHL, B-cell non-Hodgkin lymphoma; HCL, hairy cell leukemia; MOA, mode of action; PCD, programmed cell death.
A key concept that may be obvious to the practicing hematologist/oncologist but is nevertheless worthy of attention is that mAbs generally exhibit limited efficacy as single agents. For example, the response rate of newly diagnosed diffuse large B-cell lymphoma (DLBCL) to rituximab alone is up to 37%,4 to the standard chemotherapy backbone cyclophosphamide, doxorubicin hydrochloride (hydroxydaunorubicin), vincristine sulfate (Oncovin), and prednisone (CHOP) is 69%, and to rituximab combined with CHOP is 83%.5 In chronic lymphocytic leukemia (CLL), rituximab alone, fludarabine with cyclophosphamide, and fludarabine with cyclophosphamide with rituximab lead to complete response (CR) rates of 4 to 19, 22, and 44%, respectively, and a median progression-free survival of 19-43, 32.9, and 56.8 months.25-27 Similarly, daratumumab monotherapy in relapsed/refractory multiple myeloma (R/R MM) confers an overall response rate (ORR) of 29.2%, whereas daratumumab combined with bortezomib and dexamethasone is 82.9%.28,29 These observations are particularly noteworthy in the context of the mechanism of action of these mAbs, where one would a priori expect that the combination with immunosuppressive chemotherapy should be less than additive. This paradox remains, in our view, relatively unexplained with the exception of some literature on immunogenic cell death (ICD) and depletion of immunosuppressive cells by certain chemotherapeutic agents.30-32 Interestingly, the addition of chemotherapy can also improve the antitumor response of immune checkpoint blockade,33 despite the detrimental effect of chemotherapy on the immune system (lymphopenia). A possible explanation is the induction of an adaptive immune response by ICD of tumor cells, which enhances priming and activation of cytotoxic T cells (Figure 1).
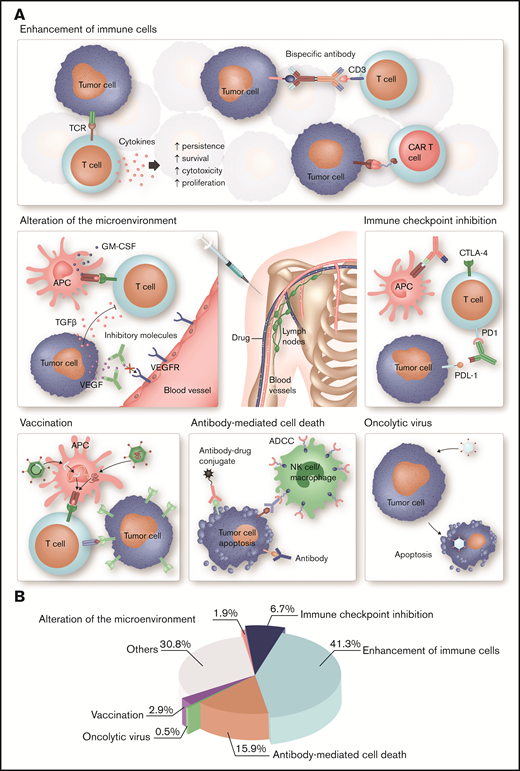
Different strategies of immunotherapy in hematology. (A) Each type of immunotherapy differently affects the immune system. ADC, OV, and T cells engineered with a tumor antigen-specific TCR or CAR directly mediate an antitumor response, whereas cytokines, bispecific antibodies, immune checkpoint blockade, and therapeutic cancer vaccines stimulate endogenous immune pathways and thus indirectly induce a therapeutic effect. More specifically, immune stimulatory cytokines, such as IL-2 and interferon-α, are used to enhance the proliferation, cytotoxicity, persistence, and survival of T cells. Bispecific antibodies function as essential link between tumor cells and T cells mediating T-cell activation and tumor cell lysis. Another strategy to enhance tumor cell recognition is the ex vivo engineering of patient T cells with a CAR. Immune evasion is a common feature of tumor cells. Antibodies targeting immune checkpoints can prevent exhaustion of cytotoxic T cells and thus improve antitumor immunity. OV specifically infects tumor cells, which result in ICD. The binding of mAbs to tumor cells leads to activation of the innate immune system and tumor cells destruction. An advancement is ADCs, which specifically bind to tumor cells and induce cell death after internalization because of their cytotoxic conjugate. Therapeutic cell-based, peptide-based, or gene-based cancer vaccines induce tumor-antigen presentation by APC to boost a specific and long-lasting antitumor immune response. Antibodies or small molecules are used to neutralize immunosuppressive, tumor-derived soluble factors, such as TGF-β, IL-10, or VEGF, and thus ameliorate antitumor immunity. (B) Articles published in Blood Advances on immunology and immunotherapy in the period from November 2016 to April 2020 were classified as indicated. Most of these articles focus on cell-based immunotherapies, ICIs, and mAbs, which is in accordance with their major clinical relevance in hematology. GM-CSF, granulocyte-macrophage colony-stimulating factor; TGF-β, transforming growth factor-β; VEGF, vascular endothelial growth factor. Professional illustration by Somersault18:24.
Different strategies of immunotherapy in hematology. (A) Each type of immunotherapy differently affects the immune system. ADC, OV, and T cells engineered with a tumor antigen-specific TCR or CAR directly mediate an antitumor response, whereas cytokines, bispecific antibodies, immune checkpoint blockade, and therapeutic cancer vaccines stimulate endogenous immune pathways and thus indirectly induce a therapeutic effect. More specifically, immune stimulatory cytokines, such as IL-2 and interferon-α, are used to enhance the proliferation, cytotoxicity, persistence, and survival of T cells. Bispecific antibodies function as essential link between tumor cells and T cells mediating T-cell activation and tumor cell lysis. Another strategy to enhance tumor cell recognition is the ex vivo engineering of patient T cells with a CAR. Immune evasion is a common feature of tumor cells. Antibodies targeting immune checkpoints can prevent exhaustion of cytotoxic T cells and thus improve antitumor immunity. OV specifically infects tumor cells, which result in ICD. The binding of mAbs to tumor cells leads to activation of the innate immune system and tumor cells destruction. An advancement is ADCs, which specifically bind to tumor cells and induce cell death after internalization because of their cytotoxic conjugate. Therapeutic cell-based, peptide-based, or gene-based cancer vaccines induce tumor-antigen presentation by APC to boost a specific and long-lasting antitumor immune response. Antibodies or small molecules are used to neutralize immunosuppressive, tumor-derived soluble factors, such as TGF-β, IL-10, or VEGF, and thus ameliorate antitumor immunity. (B) Articles published in Blood Advances on immunology and immunotherapy in the period from November 2016 to April 2020 were classified as indicated. Most of these articles focus on cell-based immunotherapies, ICIs, and mAbs, which is in accordance with their major clinical relevance in hematology. GM-CSF, granulocyte-macrophage colony-stimulating factor; TGF-β, transforming growth factor-β; VEGF, vascular endothelial growth factor. Professional illustration by Somersault18:24.
Subsequently, antibody-drug conjugates (ADCs) were developed to capitalize on the property of some lineage-associated proteins to internalize upon cross-linkage, thereby delivering chemotherapy, radiation, or biologic toxins into the target cell and causing relatively specific cell death. We do not consider these drugs a form of immunotherapy because the effector mechanism is that of the toxin conjugate. However, ADCs generally have more impressive single-agent activity than naked mAbs (Table 2).
Therapeutic efficacy of naked mAb compared with ADC
| Target | Naked mAb | ADC |
|---|---|---|
| BCMA | No data available | Belantamab |
| Cytotoxic payload | Monomethylauristatin F (MMAF) | |
| Indication/status | R/R MM (FDA approved) | |
| Median PFS (mo) | 2.9 | |
| 1-y PFS | n.d. | |
| ORR, % | 31 | |
| Reference Clinical trial | 16 NCT03525678 | |
| CD19 | Tafasitamab | Loncastuximab |
| Cytotoxic payload | Pyrrolobenzodiazipine (PBD) | |
| Indication/status | R/R B-NHL (FDA approved) | R/R B-NHL (FDA approved) |
| Median PFS (mo) | 2.7 (DLBCL) 8.8 (FL) | 5.5 |
| 1-y PFS, % | 39 | n.d. |
| ORR, % | 29 | 59.4 (CR 24.1%) |
| Reference Clinical trial | 34 NCT01685008 | 35 NTC02669017 |
| CD20 | Rituximab | Ibritumomab |
| Cytotoxic payload | 90Y | |
| Indication/status | R/R B-NHL (FDA approved) | R/R B-NHL (FDA approved) |
| Median PFS (mo) | 10.1 | 11.2 |
| 1-y PFS | n.d. | n.d |
| ORR, % | 56 | 80 |
| Reference | 17 | 17 |
| CD22 | Epratuzumab | Inotuzumab |
| Cytotoxic payload | Calicheamicin | |
| Indication/status | R/R ALL (phase 2 study) | R/R ALL (FDA approved) |
| Median OS, mo | 3 | 7.7 |
| 1-y PFS | n.d. | n.d. |
| ORR, % | 50 | 80.7 CR |
| Reference Clinical trial | 36 NTC01219816 | 18 NCT01564784 |
| CD30 | SGN-30 | Brentuximab (FDA approved) |
| Cytotoxic payload | Monomethylauristatin E (MMAE) | |
| Indication/status | R/R HL (phase 2 study) | R/R HL |
| ORR, % | 0 (28.9% stable disease) | 86 |
| Median PFS | n.d. | n.d. |
| 2-y PFS | n.d. | 82.1% |
| Reference Clinical trial | 37 | 38 NCT01712490 |
| CD33 | Lintuzumab | Gemtuzumab |
| Cytotoxic payload | Calicheamicin | |
| Indication/status | R/R AML (phase 3 study, failed) | R/R AML (FDA approved) |
| ORR, % | 36 | 38.8 |
| Median PFS | Median survival 171.5 d | 1 y RFS 18.5% |
| Median OS | n.d. | 1 y OS 26% |
| Reference Clinical trial | 39 NCT00006045 | 40 |
| CD79b | No clinical data available | Polatuzumab |
| Cytotoxic payload | Monomethylauristatin E (MMAE) | |
| Indication/status | R/R DLBCL (FDA approved) | |
| Median PFS | 9.5 mo | |
| 1-y PFS | n.d. | |
| CR, % | 40 | |
| Reference Clinical trial | 24 NCT02257567 |
| Target | Naked mAb | ADC |
|---|---|---|
| BCMA | No data available | Belantamab |
| Cytotoxic payload | Monomethylauristatin F (MMAF) | |
| Indication/status | R/R MM (FDA approved) | |
| Median PFS (mo) | 2.9 | |
| 1-y PFS | n.d. | |
| ORR, % | 31 | |
| Reference Clinical trial | 16 NCT03525678 | |
| CD19 | Tafasitamab | Loncastuximab |
| Cytotoxic payload | Pyrrolobenzodiazipine (PBD) | |
| Indication/status | R/R B-NHL (FDA approved) | R/R B-NHL (FDA approved) |
| Median PFS (mo) | 2.7 (DLBCL) 8.8 (FL) | 5.5 |
| 1-y PFS, % | 39 | n.d. |
| ORR, % | 29 | 59.4 (CR 24.1%) |
| Reference Clinical trial | 34 NCT01685008 | 35 NTC02669017 |
| CD20 | Rituximab | Ibritumomab |
| Cytotoxic payload | 90Y | |
| Indication/status | R/R B-NHL (FDA approved) | R/R B-NHL (FDA approved) |
| Median PFS (mo) | 10.1 | 11.2 |
| 1-y PFS | n.d. | n.d |
| ORR, % | 56 | 80 |
| Reference | 17 | 17 |
| CD22 | Epratuzumab | Inotuzumab |
| Cytotoxic payload | Calicheamicin | |
| Indication/status | R/R ALL (phase 2 study) | R/R ALL (FDA approved) |
| Median OS, mo | 3 | 7.7 |
| 1-y PFS | n.d. | n.d. |
| ORR, % | 50 | 80.7 CR |
| Reference Clinical trial | 36 NTC01219816 | 18 NCT01564784 |
| CD30 | SGN-30 | Brentuximab (FDA approved) |
| Cytotoxic payload | Monomethylauristatin E (MMAE) | |
| Indication/status | R/R HL (phase 2 study) | R/R HL |
| ORR, % | 0 (28.9% stable disease) | 86 |
| Median PFS | n.d. | n.d. |
| 2-y PFS | n.d. | 82.1% |
| Reference Clinical trial | 37 | 38 NCT01712490 |
| CD33 | Lintuzumab | Gemtuzumab |
| Cytotoxic payload | Calicheamicin | |
| Indication/status | R/R AML (phase 3 study, failed) | R/R AML (FDA approved) |
| ORR, % | 36 | 38.8 |
| Median PFS | Median survival 171.5 d | 1 y RFS 18.5% |
| Median OS | n.d. | 1 y OS 26% |
| Reference Clinical trial | 39 NCT00006045 | 40 |
| CD79b | No clinical data available | Polatuzumab |
| Cytotoxic payload | Monomethylauristatin E (MMAE) | |
| Indication/status | R/R DLBCL (FDA approved) | |
| Median PFS | 9.5 mo | |
| 1-y PFS | n.d. | |
| CR, % | 40 | |
| Reference Clinical trial | 24 NCT02257567 |
n.d., not determined; OS, overall survival; PFS, progression-free survival; RFS, relapse-free survival; R/R ALL, relapsed/refractory acute lymphoblastic leukemia; R/R AML, relapsed/refractory acute myeloid leukemia; R/R B-NHL, relapsed/refractory B-cell non-Hodgkin lymphoma.
Two interrelated questions arise when considering this interesting class of drugs: How specific are they really, and how can they be made even more active? The side-effect profile of gemtuzumab and inotuzumab (cytopenia and hepatotoxicity) is remarkably similar given they target distinct hematopoietic lineages, brentuximab is associated with an up to 70% incidence of peripheral neuropathy,41 and belantamab mafodotin is commonly associated with keratopathy.42 These off-target toxicities suggest that future efforts to improve response using more toxic payloads would have to be done cautiously and with a detailed understanding of the conjugation chemistry, metabolism, and pharmacokinetics of the system as a whole. Novel ADCs in early-phase clinical trials include those targeting CCR7 (CLL, NHL), CD46 (R/R MM), CD71 (R/R AML, R/R ALL), CD74 (advanced B-cell malignancies), CD123 (R/R AML, R/R ALL), and ROR1 (hematologic cancer) (www.clinicaltrials.gov, searched on 12 January 2021).
mAbs have also been studied for the treatment of hematologic autoimmune diseases, particularly ones thought to be driven by autoreactive antibodies. Depletion of B cells with rituximab is surprisingly effective in some forms of autoimmune hemolytic anemia, immune thrombocytopenia, immune-mediated thrombotic thrombocytopenic purpura, treatment of inhibitors in hemophilia, and perhaps in chronic graft-versus-host disease (GVHD).43-47 With some exceptions, this approach is not particularly mechanistically driven (perhaps more opportunistic, given the widespread availability and comfort level on the part of clinicians with the use of rituximab) because most autoantibodies are likely produced by plasma cells, and indeed, in some of the abovementioned disorders (such as GVHD), the pathogenic process remains quite opaque. Furthermore, mAbs as specific neutralizers are of great interest in immune diseases that are driven primarily by the production of a cytokine, such as anti-interleukin-5 (IL-5) (mepolizumab) in hypereosinophilic syndrome,48 anti-IL-6 (siltuximab) in Castleman disease,49 and anti-interferon-γ (emapalumab) in hemophagocytic lymphohistiocystosis (HLH).50 A different method of cytokine inhibition is cytokine receptor blockade, such as IL-6R (tocilizumab; studied in the treatment of rheumatoid arthritis,51 cytokine release syndrome [CRS],52 and COVID-1953 ) or of the high-affinity IL-2 receptor (basiliximab; studied in solid-organ transplantation54 and GVHD55 ).
Alternative multispecific antigen-targeting formats, such as bispecific T-cell engaging antibodies (BiTEs), are discussed in a subsequent section because their major mechanism of action is that of the recruited effector cell.
Immune checkpoint blockade or activation
T-cell activation is a rigorously controlled process that is dependent on signals provided by the T-cell receptor (TCR) complex and its interaction with regulatory proteins. A dynamic interplay with inhibitory and stimulatory proteins modulates the degree of T-cell activation to allow tolerance to self-antigens (inhibitory) while initiating an adaptive immune response to foreign antigens (stimulatory). Immune checkpoints are essential inhibitory stimuli, which normally maintain immune responses within a desired physiologic range by a temporary downregulation of T-cell function.
Malignant cells often coopt this protective mechanism and evade the host immune system by expressing the ligands of immune checkpoint pathways, such as CTLA-4 and PD-1 pathways. Blockade of receptors or ligands involved in these inhibitory mechanisms can in some cases reverse the tumor-mediated downregulation of T-cell function and enhance an effective antitumor immune response at the priming (CTLA-4) or tissue effector (PD-1) phase.56,57 Immunologic tolerance is retained through clonal deletion of self-reactive clones during negative selection in the thymus (central tolerance) or through peripheral tolerance, a loosely defined process that includes cell-intrinsic (eg, induction of anergy) and cell-extrinsic (regulatory T [Treg] cells, myeloid-derived suppressor cells) mechanisms. Blockade of inhibitory immune checkpoints can activate otherwise exhausted antitumor T cells. One drawback of immune checkpoint blockade is the greater probability for the activation of autoreactive T-cell clones with low signal strength that are normally incapable of generating an effective immune.58 Therefore, patients with preexisting active autoimmune disorders have historically been excluded from clinical trials using immune checkpoint inhibitors (ICIs) because of the susceptibility to develop severe adverse effects. However, mounting evidence supports the safety and effectiveness of immune checkpoint blockade in this group of patients.59
The first promising clinical results with checkpoint blockade therapy were in the treatment of solid tumors, especially melanoma. In this malignancy, blocking of the CTLA-4 and/or PD-1 pathway has shown superior activity with a potential to induce durable response.60,61 Today, we also know that a higher mutational burden across multiple solid tumor types correlates with a greater immunogenicity and better response to ICIs.62 Hematological malignancies, however, have a very low mutational burden, which may explain the poor responses to immune checkpoint inhibition in the treatment of these diseases.63,64 We suspect however that this is not the only answer, because response rates to ICIs in hematologic malignancies with high rates of somatic mutations, such as those with mutations in TP53, do not appear to be higher.
A notable exception is Hodgkin lymphoma (HL), where alterations in 9p24.1 JAK and MEK/ERK signaling are recurrent genetic abnormalities that lead to the overexpression of PD-1 ligands (PD-L1) on Hodgkin-Reed-Sternberg cells.65 The interaction of PD-L1 with PD-1 results in dephosphorylation of proteins involved in the TCR signaling pathway, which terminates the signaling cascade and consequently inhibits T-cell activity and proliferation. Blocking PD-1 during the effector phase restores the immune function of T cells and enhances the antitumor activity, including T-cell proliferation, cytokine production, and survival.66 In clinical studies, treatment with anti–PD-1 antibodies, nivolumab or pembrolizumab, resulted in an ORR of 87% and 65% in patients with R/R Hodgkin lymphoma (R/R HL), leading to their approval in 2016 by Food and Drug Administration (FDA).67,68
Another consideration is that the PD-1/PD-L1 axis is far from the only immune checkpoint. Following the therapeutic success of CTLA-4 and PD-1 blockade in some solid cancers, many other T-cell costimulatory molecules are now being investigated in preclinical and clinical studies. Among these molecules, LAG-3 and TIM-3 are the most advanced candidates, but there is also growing evidence for the therapeutic relevance of TIGIT and VISTA blockade to enhance antitumor immunity in hematologic disease.69,70
LAG-3 is mainly expressed on activated CD4 and CD8 T cells, and coexpression with PD-1 correlates with an exhausted phenotype. In follicular lymphoma (FL), intratumoral PD-1+ LAG-3+ T cells were reported to be functionally suppressed, and blockade of both PD-1 and LAG-3 enhanced the functionality of intratumoral T cells.71 Currently, several phase 1/2 clinical trials are ongoing to evaluate the therapeutic potential of LAG-3 antibodies as single or in combination with PD-1 inhibitors in relapsed or refractory B-cell malignancies (NCT03489369, NCT03005782, NCT02061761). In addition, a dual-targeting antibody specific for both PD-1 and LAG-3 is investigated in a phase 1 study in patients with unresectable or metastatic neoplasms, including DLBCL (NCT03219268). An acceptable safety profile and encouraging evidence of antitumor activity have already been reported for this dual-targeting antibody.72
More recently, it was demonstrated that the number of PD-1/TIM-3 double-positive T-cell subsets is increased in newly diagnosed and relapsed acute myeloid leukemia (AML) compared with healthy specimens.73 Interestingly, TIM-3 expression was also significantly elevated in leukemia stem cells (LSCs) and leukemic progenitors, but not in normal hematopoietic stem cells or progenitors.74 Given that LSCs are considered to be responsible for AML relapse after standard therapies, targeting of TIM-3 represents a promising novel approach in eliminating LSCs and preventing disease relapse. Therefore, TIM-3 targeting mAbs, as single agent or combined with anti–PD-1 antibodies, are currently tested in phase 1 clinical trials for both solid tumors and hematologic malignancies (NCT03489343, NCT03311412).
Another form of tumor immune evasion is the upregulation of CD47, which is a “don’t eat me” signal whose overexpression results in inhibition of phagocytosis by macrophages. CD47 is highly expressed in solid tumors and myeloid malignancies.75-78 In preclinical models of AML and myelodysplastic syndromes (MDS), CD47 blockade led to an enhanced antitumor response.77,78 In addition, anti-CD47 antibodies stimulate ADCP, thus enhancing priming and memory response of CD8 T cells.79 The therapeutic targeting of CD47 as a macrophage immune checkpoint is being investigated in several early clinical trial studies for the treatment of AML, non-Hodgkin lymphoma (NHL), MDS, HL, MM, and multiple solid tumors. A phase 1 trial of an anti-CD47 antibody in combination with azacytidine, a hypomethylating and cytotoxic agent, led to objective responses in 64% of AML patients and 92% in MDS patients with 55% or 50% achieving CR, respectively80 (NCT03248479). Although the patient number and follow-up time are limited, this report is encouraging and proves the clinical applicability of macrophage checkpoint blockade.
Although blocking antibodies have transformed cancer therapy, the development of agonist antibodies that activate costimulatory receptors to amplify antitumor immunity has been less effective. After the disastrous outcome of a CD28 superagonist antibody, which caused massive CRS and multiorgan failure in 6 healthy individuals due to T-cell activation without TCR engagement,81 current studies focus on targeting of receptors that are upregulated following T-cell activation, such as 4-1BB, OX40, GITR, CD27, and ICOS. A combination study with rituximab and utomilumab, a 4-1BB activating mAb, in 67 patients with R/R follicular lymphoma (R/R FL) and other CD20+ NHL found a 21.2% objective response rate with 4 complete and 10 partial responses. Importantly, no patient experienced dose-limiting toxicity.82 Currently, utomilumab is being tested in combination with avelumab (anti–PD-L1 mAb) in DLBCL (NCT02951156). A phase 1 study evaluated the dosage and safety of an anti-CD27 agonist antibody (varlilumab) in patients with hematologic malignancies and solid tumors (NCT01460134). In addition to T-cell stimulation, varlilumab mediates direct lysis of CD27+ lymphoma cells. However, varlilumab treatment only led to a CR in one of 10 HL patients and no objective response in 18 NHL patients (3 patients with stable disease).83 Because of this modest single-agent effect, varlilumab is further studied in combination with rituximab (NCT03307746). In this approach the induction of CDC and ADCC by rituximab is thought to be complemented by the CD27 mAb-mediated costimulation during T-cell activation.83 There are still many open questions on dosing and scheduling of agonistic antibodies, antibody structure, and combination with ICIs to improve treatment efficiency, reduce toxicity, and prevent ADCC, exhaustion, and activation-induced cell death following overstimulation of T cells with activating antibodies.
Blockade of costimulatory molecules or activation of inhibitory signaling is also of great interest for the treatment of auto- or alloimmunity. For instance, abatacept and belatacept, synthetic CTLA-4–Ig fusion proteins, are used to treat rheumatoid arthritis and improve graft survival after organ transplantation.84 An ongoing phase 2 clinical trial (NCT01743131) is investigating the addition of abatacept for the prevention of GVHD in blood cancer patients undergoing stem cell transplant. Early data showed that 6.8% of patients treated with abatacept developed severe acute GVHD compared with 14.8% in the standard treatment cohort. Importantly, abatacept addition did not increase infection risk or increase the relapse incidence. In addition, abatacept treatment was associated with a severe acute GVHD free-survival benefit (97.7% vs 58.5%).85,86 Based on these results, the FDA named abatacept a breakthrough therapy for the prevention of GVHD in hematopoietic stem cell transplants (HSCTs) from unrelated donors.
Enhancement of immune cells
In addition to the above approaches, immune cell activity may be augmented by increasing the number of cells (stimulating proliferation ex vivo followed by adoptive transfer; administration of homeostatic or activating cytokines) or endowing them with novel functions or antigen specificity.
Ex vivo stimulation and expansion using activating beads and cytokines have been applied to effector T cells, Treg cells, and natural killer (NK) cells,87-89 whereas effector T cells have also been stimulated ex vivo with antigen-presenting cells (APCs) bearing tumor-associated or viral antigens.90-92 For the treatment of drug-refractory virus-driven cancers as well as viral infections in patients undergoing HSCT, in vitro sensitization of virus-specific T cells with viral-infected APCs or APCs loaded with infected cell lysates or synthetic peptides represents an effective therapeutic alternative. The efficiency of autologous or allogeneic Epstein-Barr virus–, cytomegalovirus-, or human papilloma virus-specific T-cell therapies is currently investigated in clinical trials, including NCT02379520, NCT02973113, and NCT03475212. Virus-specific T cells are also a potential source of allogeneic T cells for transduction with a chimeric antigen receptor (CAR). Because of the native virus-specific TCR, Epstein-Barr virus– or cytomegalovirus-specific CAR T cells have the potential of an enhanced in vivo proliferation while lacking alloreactive potential.93,94 Another interesting approach is the generation of cytokine-activated memory-like (ML) NK cells. Following ex vivo stimulation with IL-12, IL-15, and IL-18, NK cells exhibit memory-like characteristics with enhanced antitumor activity. A first-in-human phase 1 clinical trial revealed that 5 of 9 AML patients responded to adoptively transferred ML NK cells, including 4 complete remissions.95 In a second clinical trial, patients with R/R AML received a haploidentical hematopoietic cell transplant followed by same-donor ML NK cells to avoid elimination of ML NK cells by recipient allogeneic immune response. The first patients treated showed an efficient expansion and persistence (≥2 months) of highly functional ML NK cells providing first evidence for a long-term response96 (NCT02782546).
Cytokines can also be administered directly in vivo either to stimulate antitumor or to activate Treg cells.97-99 However, cytokines are typically promiscuous in their activity. IL-2 was initially developed to stimulate effector lymphocytes but is now known to stimulate Treg cells even more potently. Interesting new developments that have, as yet, to make their way into the hematologic malignancy space include synthetic cytokine/cytokine receptor pairs whose specificity can be tailored to desired cell populations. For instance, T cells engineered to express synthetic IL-2 and IL-2R, which interact with each other but not with endogenous IL-2 or IL-2R, selectively expanded and promoted an antitumor response in preclinical melanoma models.100
Another approach is the design of chimeric antibody-cytokine fusion proteins (immunocytokines), which comprise a tumor-specific single-chain variable fragment (scFv) conjugated to an immunostimulatory cytokine, such as IL-2, IL-15, or tumor necrosis factor-α, to improve the local accumulation and pharmacokinetics compared with the native cytokines.101,102 Clinical phase 2/3 studies are currently testing the safety and activity of immunocytokines as monotherapy or in combination with ICIs for the treatment of solid tumors (NCT03420014, NCT03567889). Additional clinical investigations combine cytokines with anticancer vaccines, ICIs, and antibody-based therapies.103,104 For example, in NHL patients, recombinant IL-21 has been tested in combination with rituximab, achieving clinical response in 8 out of 19 patients.105 Further studies are currently recruiting patients to investigate the combination of recombinant IL-15 and CD20-targeting antibody therapy for the therapy of CLL (NCT03759184) or the synergistic effect of IL-15 and ICIs in relapsed/refractory mature T-cell malignancies (NCT03905135).
BiTEs can be thought of as a modality to endow effector cells with novel specificity. To date, most approaches use an anti-CD3ε antibody fragment as T-cell engaging domain fused to a scFv targeting a tumor-associated antigen. Binding of the BiTE to both targets, the TCR complex and the tumor antigen, mediates the formation of a cytolytic synapse resembling natural immunological synapses. Thus far, blinatumomab, a CD3×CD19 BiTE, is the only BiTE with FDA approval and is used for the treatment R/R B-cell precursor ALL (pre–B-ALL). Although blinatumomab showed only sustained responses, impressive results in phase 1 and 2 studies for R/R DLBCL, benefits in response duration have been reported and are further investigated in ongoing phase 1/2 trials.106 At this time, blinatumomab is not approved for NHL because of a lack of phase 3 trials, difficult mode of administration, and availability of alternative options with similar therapeutic efficacy.107 Notably, blinatumomab is particularly active in ALL patients with the minimal residual disease where its CR rate is 80%, whereas in the setting of active disease, the CR rate is 43%.108,109 Not surprisingly, blinatumomab is now being tested in combination with ICIs. One study of blinatumomab in combination with pembrolizumab reported an acceptable toxicity and a 50% ORR in adults with heavily pretreated R/R B-ALL.110
To improve the feasibility and/or efficacy of the BiTE platform, novel CD19×CD3 constructs have been developed with improved half-life and higher CD3 affinity. Two candidates, AFM11 and MGD011, have been tested in patients with NHL and CLL or NHL and ALL, but their clinical development was discontinued because of high levels of neurotoxicity.111,112 An alternative target in B-cell malignancies is CD20, and several CD20×CD3 BiTEs are tested in ongoing clinical trials (Table 3). In an early-phase clinical trial, treatment with single-agent mosunetuzumab, a fully humanized CD20×CD3 bispecific antibody, induced durable responses in patients with B-cell NHL, even in those who relapsed following CAR T-cell therapy.113 Treatment-associated adverse events were similar to those typically observed with CAR T-cell therapy with CRS in 28.9% and neurologic toxicity in 43.7% of patients.
Overview of immune cell redirecting bispecific antibodies investigated in ongoing clinical trials for hematologic malignancies
| Target | Molecule/drug | Condition | Phase; status; NCT# |
|---|---|---|---|
| BCMA + CD3 | JNJ-64007957 (Teclistamab) | MM | 1; recruiting; NCT03145181 1; recruiting; NCT04696809 2; recruiting; NCT04557098 |
| BMCA + CD3 | PF-06863135 (Elranatamab) | MM | 1; recruiting; NCT03269136 2; recruiting; NCT04649359 |
| BCMA + CD3 plus GPRC5D + CD3 | JNJ-64007957 (Teclistamab); JNJ-64407564 (Talquetamab) | MM | 1; recruiting; NCT04108195 1; recruiting; NCT04586426 |
| BCMA + CD3 | TNB-383B | MM | 1; recruiting; NCT03933735 |
| BCMA + CD3 | REGN5458 | MM | 1/2; recruiting; NCT03761108 |
| BCMA + CD3 | REGN5459 | MM | 1; recruiting; NCT04083534 |
| CD19 + CD3 | TNB-486 | B-cell lymphoma, FL | 1; recruiting; NCT04594642 |
| CD20 + CD3 | REGN1979 (Odronextamab) | NHL, HL, CLL | 1; active, not recruiting; NCT02651662, NCT02290951 2; active, not recruiting; NCT03888105 |
| CD20 + CD3 | RG7828 (Mosunetuzumab) | NHL, CLL | 1; recruiting; NCT02500407, NCT03671018, NCT03677141, NCT03677154 |
| CD33 + CD3 | AMG330 | AML | 1; recruiting; NCT02520427, NCT02106091 |
| CD33 + CD3 | AMV564 | AML, MDS | 1; active, not recruiting; NCT03516591, NCT03144245 |
| CD33 + CD3 | GEM333 | AML | 1; active, not recruiting; NCT03516760 |
| CD33 + CD3 | JNJ-67371244 | AML, MDS | 1; recruiting; NCT03915379 |
| CD33 + CD3 (with IL-15 crosslinker) | 161533 TriKE | MDS, AML, ASM | 1/2; recruiting; NCT03214666 |
| CD38 + CD3 | ISB 1342 | MM | 1/2; recruiting; NCT03309111 |
| CD123 + CD3 | MGD006 (Flotetuzumab) | AML, MDS | 1; recruiting; NCT02152956 1; recruiting; NCT04681105 |
| CD123 + CD3 | APVO436 | AML, MDS | 1; recruiting; NCT03647800 |
| CD123 + CD3 | JNJ-63709178 | AML | 1; recruiting; NCT02715011 |
| CD123 + CD3 | Xmab14045 (Vibecotamab) | AML, B-cell ALL, BPDCN, CML | 1; recruiting; NCT02730312 |
| GPRC5D + CD3 | JNJ-64407564 (Talquetamab) | Hematological malignancies | 1; recruiting; NCT03399799 1; recruiting; NCT04634552 |
| Target | Molecule/drug | Condition | Phase; status; NCT# |
|---|---|---|---|
| BCMA + CD3 | JNJ-64007957 (Teclistamab) | MM | 1; recruiting; NCT03145181 1; recruiting; NCT04696809 2; recruiting; NCT04557098 |
| BMCA + CD3 | PF-06863135 (Elranatamab) | MM | 1; recruiting; NCT03269136 2; recruiting; NCT04649359 |
| BCMA + CD3 plus GPRC5D + CD3 | JNJ-64007957 (Teclistamab); JNJ-64407564 (Talquetamab) | MM | 1; recruiting; NCT04108195 1; recruiting; NCT04586426 |
| BCMA + CD3 | TNB-383B | MM | 1; recruiting; NCT03933735 |
| BCMA + CD3 | REGN5458 | MM | 1/2; recruiting; NCT03761108 |
| BCMA + CD3 | REGN5459 | MM | 1; recruiting; NCT04083534 |
| CD19 + CD3 | TNB-486 | B-cell lymphoma, FL | 1; recruiting; NCT04594642 |
| CD20 + CD3 | REGN1979 (Odronextamab) | NHL, HL, CLL | 1; active, not recruiting; NCT02651662, NCT02290951 2; active, not recruiting; NCT03888105 |
| CD20 + CD3 | RG7828 (Mosunetuzumab) | NHL, CLL | 1; recruiting; NCT02500407, NCT03671018, NCT03677141, NCT03677154 |
| CD33 + CD3 | AMG330 | AML | 1; recruiting; NCT02520427, NCT02106091 |
| CD33 + CD3 | AMV564 | AML, MDS | 1; active, not recruiting; NCT03516591, NCT03144245 |
| CD33 + CD3 | GEM333 | AML | 1; active, not recruiting; NCT03516760 |
| CD33 + CD3 | JNJ-67371244 | AML, MDS | 1; recruiting; NCT03915379 |
| CD33 + CD3 (with IL-15 crosslinker) | 161533 TriKE | MDS, AML, ASM | 1/2; recruiting; NCT03214666 |
| CD38 + CD3 | ISB 1342 | MM | 1/2; recruiting; NCT03309111 |
| CD123 + CD3 | MGD006 (Flotetuzumab) | AML, MDS | 1; recruiting; NCT02152956 1; recruiting; NCT04681105 |
| CD123 + CD3 | APVO436 | AML, MDS | 1; recruiting; NCT03647800 |
| CD123 + CD3 | JNJ-63709178 | AML | 1; recruiting; NCT02715011 |
| CD123 + CD3 | Xmab14045 (Vibecotamab) | AML, B-cell ALL, BPDCN, CML | 1; recruiting; NCT02730312 |
| GPRC5D + CD3 | JNJ-64407564 (Talquetamab) | Hematological malignancies | 1; recruiting; NCT03399799 1; recruiting; NCT04634552 |
ASM, advanced systemic mastocytosis; BPDCN, blastic plasmacytoid dendritic cell neoplasm; CML, chronic myeloid leukemia; GPRC5D, G-protein–coupled receptor family C group 5 member D; TriKE, trispecific killer engager.
In the treatment of MM, daratumumab and belantamab have been shown to be effective; therefore, several BiTE constructs targeting CD38 or BCMA are currently evaluated in early-phase clinical trials (Table 3). An interesting construct in the treatment of HL is AFM13, a tetravalent bispecific antibody activating the innate immune system (CD30×CD16). In a phase 1 trial, 3 out of 26 patients with R/R HL achieved a partial response and 13 patients achieved a stable disease.114 The antibody was well tolerated (grade 3 AE 9%) and tested in phase 2 trials in R/R HL (NCT02321592), cutaneous lymphoma (NCT03192202), and in combination with pembrolizumab in R/R HL (NCT02665650).
In AML, several BiTEs have been developed, and phase 1 trials with CD33×CD3 or CD123×CD3 are ongoing (Table 3). First reports of flotetuzumab, a CD123×CD3 bispecific antibody-derived molecule, in patients with AML and high-risk MDS, showed a significant increase of CD8 T cells in bone marrow samples and an ORR of 43% with manageable toxicity (grade ≥3 CRS 13%)115 (NCT02152956).
Remarkably, the reasons for primary or secondary failure of BiTEs are relatively underexplored. Therefore, combining BiTE with ICIs may play a key role in further advancing BiTE therapy. Another tumor-resistance mechanism is the downregulation of targeted antigens. Loss of CD19 expression following blinatumomab treatment is reported in 8% to 30% of relapsed ALL patients.116,117 This issue could be addressed by the design of multivalent BiTE molecules, which enhance target avidity as well as trispecific antibodies that recognize >1 tumor antigen.118
Novel functions can be conferred on effector T cells, NK cells, or macrophages through genetic means. T cells can be engineered to express a transgenic TCR specific for a tumor-associated antigen in the context of the relevant major histocompatibility (MHC) molecule or a CAR specific for a cell-surface target. Most tumor-driving mutations or overexpressed tumor-associated antigens are located intracellularly and therefore are not accessible to CAR T cells. However, the major limitations to date of TCR transgenic T cells are the requirement for MHC presentation and matching (particularly important because MHC downregulation is a common tumor immune evasion mechanism), technical difficulties with expression of costimulatory molecules, and the need to replace or delete the endogenous α/β TCR chains in order to reduce the chance of mispairing leading to novel (and potentially pathogenic) specificities.119,120
To date, 4 CD19-specific CAR T-cell therapies, axicabtagene ciloleucel, tisagenlecleucel, brexucabtagene autoleucel, and lisocabtagene maraleucel, have been approved for the treatment of B-ALL and certain B-cell lymphoma,121-125 and 1 B-cell maturation antigen (BCMA)-directed CAR T-cell product (idecabtagene vicleucel) for MM.126 Numerous clinical trials are currently ongoing to investigate CAR T cells for the treatment of hematologic neoplasms. The most promising targets for these studies are CD19, CD20, CD22, and BCMA.127 TCR T-cell products have not yet been approved, but clinical trials are evaluating the safety and therapeutic activity of HA-1 and WT1-specific TCR T cells in AML, MDS, and ALL patients (NCT03326921, NCT02770820).
Although CAR T-cell therapy is approved by the FDA as standard of care for some forms of aggressive, relapsed, or refractory hematologic malignancies, there are several challenges and hurdles that must be overcome to enable the widespread use of CAR T-cell therapy. Key challenges that we think are reasonably well understood include CRS, antigen escape, and manufacturing logistics. Important challenges that we think remain poorly understood in the hematologic malignancies space include neurotoxicity, trafficking, interactions with the tumor microenvironment, and limited in vivo persistence.128,129 Preclinical and clinical studies are addressing this wide range of obstacles in order to broaden the applicability of CAR T-cell therapy. The risk of side effects, including CRS and neurotoxicity, correlates with tumor burden, the dose of infused CAR T cells, CAR design, and patient factors, such as age and preexisting comorbidities. The understanding of CAR-mediated pathophysiology is increasing, and predictive models for the detection and prevention of CRS or neurotoxicity are currently developed but require further validation. So far, toxicity management includes supportive care and immunosuppression with tocilizumab and sometimes corticosteroids.130 Experimental approaches to reduce toxicity and enhance the safety of CAR T-cell therapy include prevention of on-target/off-tumor effects by selecting appropriate target antigens or controlling CAR activity by suicide genes or switch-off designs. In the case of CD19-specific CAR T cell, the targeting results in healthy B cells and subsequent B-cell aplasia and hypogammaglobulinemia, which may require administration of IV immunoglobulin.131,132 Diminished proliferation, persistence, and antitumor capacity can be enhanced by engineering T cells with a less-differentiated phenotype.133 One attempt to address antigen escape and tumor heterogeneity is the combinatorial targeting of multiple antigens by BiTE-secreting CAR T cells or multispecific CAR T cells.134,135 Another well-known disadvantage of autologous CAR T-cell therapy is the costly and time-consuming manufacturing process leading to treatment delay, which is particularly problematic for patients with highly proliferative diseases. Current research uses novel gene-editing tools, such as TALEN and CRISPR-Cas9, to further refine adoptive cell therapy approaches. Such genetic tools for overexpression (viral or transposon based) or deletion (CRISPR, TALEN, or ZFN) of selected molecules allow the depletion of immune checkpoints to enhance antitumor activity or replacement the endogenous TCR with a tumor-specific TCR or CAR for the generation of allogeneic, off-the-shelf cell products.136-138 Although gene editing holds immense potential, limitations for clinical application include off-target editing events, low efficiency, and impaired expansion of ex vivo edited cells. Furthermore, most gene-editing tools have only been tested in vitro, and an open question is the safety and efficacy in clinical settings.139 Therefore, the combination of already approved drugs, such as ICIs and adoptive cell therapy, holds promise for more rapid clinical implementation. For instance, a small, single-center study at Children’s Hospital of Pennsylvania reported an improved persistence of CD19-CAR T cells because of the addition of PD-1 blockade in children with heavily pretreated B-ALL, including allo-SCT.140 Other studies testing the combination of ICIs with CAR T cells are in progress, and results are eagerly awaited. An alternative approach to enhance the efficacy and persistence of CAR T cells is the coexpression of cytokines, such as IL-12 or IL-15, or cytokine receptors that stimulate T-cell proliferation.141-143 An ongoing phase 1 trial investigates the safety and dose of CD19 CAR T cells coexpressing membrane-bound IL-15 in patients with lymphoma (NCT03579888).
In addition to therapeutic applications in oncology, bispecific antibodies and cell-based therapies have been developed for the treatment of inflammatory disorders. In 2017, the bispecific antibody emicizumab was approved for the treatment of acquired hemophilia A, a severe bleeding disorder caused by inhibitory autoantibodies to coagulation factor VIII. Emicizumab bridges activated factor IX and factor X to restore the function of missing activated factor VIII, which is needed for effective hemostasis.144 The CAR concept has also been translated to treat transplant rejection and autoimmunity. Studies reported that CAR-expressing Treg cells mediated a therapeutic benefit in mouse models of colitis, multiple sclerosis, GVHD, and islet and skin transplantation.145-147 Recently, the safety of CAR-modified Treg cells was shown in phase 1 clinical trials.148,149 Another interesting approach is the replacement of the traditional extracellular scFv with an autoantigen to target autoreactive B cells with engineered T cells, which may be a strategy to treat antibody-mediated autoimmune disease.150
Vaccination
Despite the tantalizing potential of vaccines that is clearly illustrated in the world of infectious diseases,151 including the recent severe acute respiratory syndrome coronavirus 2 vaccine,152 cancer cells are not foreign microorganisms, and the development of effective cancer vaccines still represents a major challenge for the field.
To date, only 2 therapeutic cancer vaccines have been approved by the FDA, sipuleucel-T (Provenge) and Bacillus Calmette-Guerin, which are indicated for the treatment of metastatic castration-resistant prostate cancer or nonmuscle-invasive bladder cancer, and unfortunately, show marginal efficacy.153,154
A major focus of the development of therapeutic cancer vaccines is the selection of optimal target antigens, which are aberrantly expressed self-antigens. Because high-affinity T cells that recognize self-antigens are deleted at an early stage of lymphoid cell development (central tolerance), therapeutic cancer vaccines face the challenge of activating any remaining, low-affinity T cells. New strategies are developed for the selection of more immunogenic tumor-associated self-antigens and neoantigens that harbor tumor-specific mutations to improve cancer vaccines. A pilot trial of a WT1-targeting multivalent heteroclitic peptide vaccine (galinpepimut-S) following autologous stem cell transplantation found a favorable safety profile and a robust CD4 and CD8 T-cell response in 16 high-risk MM patients, which led to an encouraging median progression-free survival of 23.6 months.155 A phase 1/2 clinical trial is currently recruiting patients to assess the combination of galinpepimut-S and pembrolizumab in patients with advanced cancers including AML (NCT03761914).
In therapeutic settings, a vaccine-stimulated immune response is challenged by a high tumor burden with established immunoregulatory pathways to dampen a natural immune response. Therefore, enhanced vaccine technologies include costimulatory components, such as adjuvants, cytokines, or other agents, that improve the efficacy of cancer vaccines. Delivery of an antigen without appropriate costimulator can result in T-cell ignorance, T-cell anergy, or even T-cell deletion.156 In addition, combinations with ICIs and other new drugs that reverse immunosuppression are showing promising results in preclinical studies.157,158 This success, however, has not yet been translated into clinical benefits. Although early-phase clinical trials show the feasibility and tolerability of cancer vaccines combined with ICIs, the immunologic effect is only marginal and does not lead to a significant improvement in overall survival when compared with ICI monotherapy.159-161 Further studies are needed to clarify whether this combination therapy has a synergistic effect and can improve patient outcome.
In contrast to therapeutic vaccines, prophylactic vaccines are more successful, and several vaccines have been approved to prevent hepatitis B virus and human papilloma virus infection, which are associated with liver and cervical cancer.162 To our knowledge, there are no data on vaccines to prevent virally driven hematological malignancies.
Vaccination strategies are also effective as allergen-specific immunotherapy. Administration of protein/peptide-based allergens in repeated and often escalating doses prevents disease progression or, in some cases, provides a curative therapy in individuals with allergic rhinitis or asthma. We are not aware of any work using tolerogenic vaccines in autoimmune hematological diseases.
Oncolytic virus
Oncolytic viruses (OVs) have the potential to specifically infect tumor cells and induce ICD, which may result in a potent and long-lasting antitumor response.163 In 2015, the first OV therapy, talimogene laherparepvec, was approved by the FDA for the treatment of nonresectable metastatic melanoma. Talimogene laherparepvec is a modified herpes simplex virus-1 that was genetically modified to express granulocyte-macrophage colony-stimulating factor (GM-CSF) combining virus-mediated cytotoxicity with immune stimulation.164 Hematologic malignancies, however, still represent a therapeutic challenge for OV therapy given the immune response to viral infections. Intravascular administration of OVs poses the risk of an excessive immune activation with CRS, systemic inflammatory response syndrome, and multiorgan failure. In the other extreme, there is the potential for rapid clearance of OVs and an ineffective dosing after intravascular delivery, illustrating why intratumoral delivery is commonly used for OVs.165,166 In addition, not all viruses are suitable for treatment of hematological neoplasms. Adenovirus, which is one of the most studied OVs, is described to be unable to lyse white blood cells,167 and many humans have neutralizing antibodies against different adenovirus serotypes, which makes a systemic administration ineffective.168,169 An interesting approach is the combination of an OV with CAR T cells to overcome the heterogeneous tumor antigen expression in solid tumors. Park et al designed an OV encoding truncated CD19, which led to a specific and stable target expression and enhanced tumor cell killing following treatment with CD19-specific CAR T cells in preclinical models.170
To date, only a few trials on OV for cancer immunotherapy have been published.171 A search from clinicaltrials.gov (14 October 2020) found 38 actively recruiting studies for OV therapy in solid tumors, but only 1 phase 1 study in hematological neoplasms that currently evaluates the combination of PD-1 blockade and OV therapy in in MM (NCT03605719).
Concluding remarks and the contributions made by Blood Advances to immunotherapy in hematology
Immunotherapy has dramatically changed the quality of life and survival of some patients. Despite rapid advances in the last decade, in our view, the field remains in an “exponential growth phase” with a ferment of basic and translational research that will likely validate the promise and help us to navigate the perils of immunotherapy (see information box below).
The holy grail of immunotherapy is as a “one-and-done” intervention, if we can discover which keys to press in order to activate the patient’s faltering endogenous immune system. However, real-world examples of this in hematology are essentially limited to long-term disease-free survival in some patients with B-cell malignancies who received a single infusion of CAR T cells directed against CD19,172 and the biological and clinical correlates of even this small group of patients remain frustratingly opaque. Short of this lofty goal, immunotherapy can and does provide an additional weapon in the therapeutic arsenal for patients whose disease has not responded to more “traditional” measures, because the toxicity and vulnerability profiles are often nonoverlapping with those of chemotherapy, radiation, surgery, transplantation, or immunosuppressants. For example, antitumor mAbs are exquisitely specific and typically well tolerated (although exhibit limited single-agent efficacy), leading to fruitful combinations with chemotherapy. Validation and experience with mAb technology paved the way to bispecific T-cell (or other effector cell) engaging antibodies, which exhibit somewhat higher single-agent activity, and to the use of mAbs to block or activate immune circuits such as the PD/PD-L1 axis. High response rates to CAR T cells in hematologic malignancies ignited efforts to genetically engineer other effector or regulatory cells in increasingly sophisticated attempts to “hack” pathobiology. The high cost of novel immunotherapies (most especially CAR T cells) is often tallied against them; however, if these therapies are curative, the high initial outlay may well prove cost-efficient in the long term because the patient would be able to avoid chronic or sequential therapy with other novel (and noncurative) agents. Another interesting observation with increasing scientific interest is the role of the gut microbiome and the response to immunotherapy and development of autoimmune disease.173,174 A study of PD-1 blockade found that a high gut microbiome diversity enhanced antitumor immune response by increased antigen presentation and improved effector T-cell functionality in the tumor microenvironment.175 However, further research is needed to identify immunostimulatory and immunosuppressive bacteria species in the gut and define their effect on responsiveness to immunotherapy. Further knowledge on the connection of the microbiome and the immune system represents a unique opportunity to activate or suppress immunity for therapeutic purposes.
The perils of immunotherapy extend beyond their well-described and increasingly well-understood side effects.176-179 We think it is crucial to understand mechanisms of response as well as failure in order to avoid expending human and material capital on novel agents that are poorly conceived or lack a therapeutic rationale. For example, PD-L1 expression is a frequent immune evasion mechanism, and blocking antibodies can enhance antitumor immunity in some solid cancers. However, other than in HL, this axis has not yet been fruitfully targeted in hematologic malignancies, and we think a sober explanation of this observation is warranted. Old dogmas should be revisited: early enthusiasm for therapeutic cytokine administration (eg, IL-2 in patients with melanoma) was dampened by negative clinical outcomes in hematology, but we now understand that IL-2 can also stimulate counterregulatory and immunosuppressive cells, and novel approaches to delivering this and other cytokines have been devised.100,142,180,181 Cancer vaccines illustrate a different peril. Vaccines are highly effective at preventing certain infectious diseases but rarely effective in treating active infections; this issue may well be more acute in the immunosuppressive cancer environment, and here, the peril of immunotherapy may be in a “type II error” wherein a potentially active treatment modality is not given the opportunity to show its full potential because it is not tested in the right setting. Indeed, AML vaccines have shown promising efficacy in patients in remission.182
Future directions in immunotherapy for hematological diseases
To understand the correlates of response and failure in order to predict which patients should receive what treatment, and to define suitable next-line therapies
To understand the mechanisms of off-target toxicity and whether these can be dissociated from efficacy
To identify cancer-specific targets to reduce on-target toxicity
Logistics and cost considerations
Rational combinations
Immunotherapy is a fast-growing field of research reflected by the large number of ongoing preclinical and clinical studies evaluating novel treatment strategies and therapeutic combinations with the aim to enhance effectivity, safety, and applicability. New findings on these topics and further research to understand the complexity of the immune system have been reported in Blood Advances in recent years, thereby contributing to the success story of immunotherapy. Most of these publications (41%) focused on the enhancement of immune cells (with two-thirds of the articles in the context of cancer therapy and one-third of the articles in the context of hyperimmunity and prevention thereof, including GVHD, HSCT, hemophilia). Sixteen percent of the articles reported novel findings on antibody-mediated immune response (three-fourths in cancer and one-fourth in autoimmunity), and 7% of the articles reported immune checkpoint inhibition. These and other developments will further clarify the role of immunotherapy in cancer and autoimmune disease within the next decade.
Acknowledgments
This work was supported in part by a Leukemia and Lymphoma Society Specialized Center of Research Grant (S.G.) and by the Mark Foundation (S.L.).
Authorship
Contribution: S.L. and S.G. wrote and revised the manuscript; both authors approved the final manuscript.
Conflict-of-interest disclosure: S.G. holds multiple patents for CAR T cell-related research. S.L. declares no competing financial interests.
Correspondence: Saar Gill, University of Pennsylvania, Smilow Center for Translational Research, Room 8-101, 3400 Civic Center Boulevard, Philadelphia, PA 19104; e-mail: saargill@pennmedicine.upenn.edu.

