

Key Points
Cleaved HK is observed in many patients with cancer, suggesting activation of the contact system.
EVs from cancer cell lines or patients with cancer express polyphosphate, bind and activate FXII, and are prothrombotic.

Abstract
Extracellular vesicles (EV) have been implicated in diverse biological processes, including intracellular communication, transport of nucleic acids, and regulation of vascular function. Levels of EVs are elevated in cancer, and studies suggest that EV may stimulate thrombosis in patients with cancer through expression of tissue factor. However, limited data also implicate EV in the activation of the contact pathway of coagulation through activation of factor XII (FXII) to FXIIa. To better define the ability of EV to initiate contact activation, we compared the ability of EV derived from different cancer cell lines to activate FXII. EV from all cell lines activated FXII, with those derived from pancreatic and lung cancer cell lines demonstrating the most potent activity. Concordant with the activation of FXII, EV induced the cleavage of high molecular weight kininogen (HK) to cleaved kininogen. We also observed that EVs from patients with cancer stimulated FXII activation and HK cleavage. To define the mechanisms of FXII activation by EV, EV were treated with calf intestinal alkaline phosphatase or Escherichia coli exopolyphosphatase to degrade polyphosphate; this treatment blocked binding of FXII to EVs and the ability of EV to mediate FXII activation. In vivo, EV induced pulmonary thrombosis in wild-type mice, with protection conferred by a deficiency in FXII, HK, or prekallikrein. Moreover, pretreatment of EVs with calf intestinal alkaline phosphatase inhibited their prothrombotic effect. These results indicate that polyphosphate mediates the binding of contact factors to EV and that EV-associated polyphosphate may contribute to the prothrombotic effects of EV in cancer.
Introduction
Circulating extracellular vesicles (EV) consisto f a heterogeneous mixture of vesicles of divergent size and origin.1 Elevated levels of EV are present in plasma from patients with cancer2-6 ; the EV may be derived from cancer and other cells in the tumor microenvironment.7-9 Characterization of EV content suggests that they may be useful cancer biomarkers.10,11
Thrombosis is a common complication of cancer, affecting up to 20% of individuals with certain cancer types.12 Patients with cancer-associated thrombosis (CAT) have increased mortality compared with patients who remain thrombosis free.13 Several mechanisms may underlie the development of CAT,14,15 including the prothrombotic effects of EV.16-20 Although studies are confounded by different methods of EV isolation and alternative approaches to assessing tissue factor antigen and/or activity,21 evidence suggests that EV tissue factor activity is associated with thrombosis in pancreatic cancer but not in other cancer types.22,23
In addition to tissue factor–mediated activation of coagulation, the contact pathway may initiate and amplify coagulation reactions.24,25 Classically, this pathway is initiated through autoconversion of factor XII (FXII) to FXIIa following binding to specific surfaces.26 Activation of FXII leads to conversion of plasma prekallikrein (PK) to plasma kallikrein (PKa), which activates additional FXII. FXIIa also activates FXI, leading to activation of the intrinsic coagulation pathway. High molecular weight kininogen (HK) is a critical cofactor for PK and FXI activation by FXIIa and is converted to cleaved HK (cHK) by PKa.27-29 Inorganic polyphosphate (polyP) released from platelet α granules and other sources may initiate contact activation and amplify coagulation reactions through its ability to activate FXII, enhance activation of FXI by thrombin, promote prothrombin cleavage, and modulate fibrin structure.30-32
There is little information available concerning the role of contact activation in CAT. One study demonstrated that EV derived from prostate cancer cells (prostasomes) activated FXII through the effects of polyP.33 Prostasomes also induced pulmonary emboli in mice in a manner inhibited by an anti-FXIIa monoclonal antibody, and they stimulated thrombin generation in normal plasma.
To further define the interactions between EV polyP from cancer cells and patients with cancer and the contact system, we examined cell lines derived from pancreas, colon, and lung cancers, as well as plasma from patients with cancer. EV derived from these cells bound FXII in a polyP-dependent manner and stimulated FXII activation with relative activity proportional to the thrombotic risk associated with each of these tumors.34 These results suggest that polyP-mediated contact activation by EV may contribute to thrombosis in cancer.
Materials and methods
Cell culture
Human dermal fibroblasts (HDFs; American Type Culture Collection) and pancreatic cancer cells (L3.6)35 were cultured in Dulbecco’s modified Eagle medium. Non–small cell lung cancer (H1975), colorectal cancer (HT29), and lymphoma (U937; all from American Type Culture Collection) cells were maintained in RPMI 1640. All media were supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin.
Isolation of EVs
Cells at 70% confluency were washed with phosphate-buffered saline (PBS) and cultured in serum-free medium for 40 hours. EV were isolated using protocols outlined by the extracellular RNA research portal (www.exRNA.org/resources/protocols). Briefly, conditioned medium was centrifuged at 450g for 10 minutes, the supernatant was recentrifuged at 2200g for 12 minutes, and the second supernatant was centrifuged at 15 000g for 30 minutes and then concentrated to 400 µL using a 50-kDa molecular weight cutoff filter (Amicon Ultra-15). The EV-containing concentrate was loaded onto a qEV size-exclusion column (SP1; Izon) equilibrated with HBS (HEPES buffered saline; 20 mM HEPES [pH 7.4] and 100 mM NaCl). Twenty-eight 500-µL fractions were collected, and the concentration of EVs in each was measured using a ZetaView NTA PMX-120 (Particle Metrix). The protein concentration in each fraction was measured using a DC Protein Assay (Bio-Rad), and fractions with the highest particle numbers and lowest protein concentrations (fractions 7-10) were pooled and concentrated. EV concentration, size distribution, and protein concentration of the pooled preparation were determined.
Isolated EV were further characterized by immunoblotting using antibodies to CD63 (sc-5275, 1:500), CD81 (sc-23962, 1:500), and CD9 (sc-13118, 1:500). Bound antibodies were detected using a horseradish peroxidase–conjugated secondary antibody (rat anti-mouse IgG1-HRP; 1144-05; Southern Biotech) and developed using SuperSignal West Pico ECL solution.
Electron microscopy
EV (10 μl) were deposited on formvar-carbon coated grids (Electron Microscopy Sciences, Hatfield, PA) for 1 hour. Grids were fixed with 4% paraformaldehyde, pH 7.4, for 10 minutes,36 washed, stained with 1% uranyl acetate, contrasted with 1.8% methylcellulose containing 0.1% uranyl acetate, and air dried. Transmission electron microscopy was performed using a Tecnai G2 Spirit BioTwin (FEI, Hillsboro, OR) at 100 kV. Digital images were obtained using an ORIUS 832 CCD 11-megapixel camera (Gatan, Pleasanton, CA).
Plasma preparation
All human studies (procurement of plasma samples) were conducted after informed written consent under an approved Cleveland Clinic Institutional Review Board protocol. Blood from healthy donors or patients with cancer was collected into vacutainer tubes containing 3.8% sodium citrate (BD Biosciences, Franklin Lakes, NJ). Platelet-poor normal human plasma (NHP) was obtained by centrifugation at 200g for 20 minutes, followed by centrifugation at 2500g for 20 minutes, and it was stored at −80°C.
FXII activation
Pooled NHP (100 μL) was added to a mixture containing 50 μL of substrate S-2302 (0.33 mM) and 50 μL of HBS, with or without EVs derived from cancer cell lines (L3.6, H1975, HT29, and U937) or HDFs, in 96-well microplates (07-200-568; Corning 3632). Dextran sulfate (DS; 10 μg/mL) was used as positive control for FXII activation. Samples were incubated for 160 minutes at 37°C, and FXIIa generation was assessed by measuring hydrolysis of S-2302 at 405 nm in a microplate reader (Bio-Tek, Winooski, VT). For relative FXIIa quantification, various concentrations of human FXIIa (Enzyme Research Laboratories) were incubated in NHP containing S-2302 to establish a standard curve. FXII-deficient plasma was obtained from a patient with inherited FXII deficiency and no detectable FXII.
To assess direct activation of FXII by EV, 60 µL of purified human FXII (375 nM; Enzyme Research Laboratories) was incubated with HBS, L3.6 EVs, and DS in 60 µL of reaction buffer (20 mM HEPES [pH 7.4], 100 mM NaCl, 0.1% polyethylene glycol 8000, 10 mM ZnCl2) containing S-2302 (330 mM). Changes in optical density at 405 nm were monitored on a microplate reader for 180 minutes. FXII cleavage was analyzed in parallel by immunoblotting using a goat anti-human FXII polyclonal antibody (Affinity Biologicals).
Cleavage of HK
Cleavage of HK as a marker of contact activation27-29 was assessed by immunoblot. Briefly, 0.25 μL of NHP, pretreated or not with cancer cell–derived EVs preincubated in the absence or presence of calf intestinal alkaline phosphatase (CIP) or corn trypsin inhibitor (CTI), was analyzed using 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis under reducing conditions. After transfer to polyvinylidene difluoride, membranes were blotted using affinity-purified rabbit antibodies raised against a peptide corresponding to a sequence in domain 5 of the human HK light chain (DHGHKHKHGHGHGKHKNKGKKN) that recognizes intact HK (∼120 kDa) and the free cHK light chain (54 kDa, 47 kDa) or antibodies raised to an epitope (CQPLGMISLMK) in the C terminus of the HK heavy chain exposed after cleavage of HK to cHK (62 kDa) (supplemental Figure 1). Bound antibodies were detected using a 1:15 000 dilution of IRDye 800CW–conjugated goat anti-rabbit immunoglobulin G (LI-COR Biosciences). The membrane was scanned using an Odyssey CLx imager, and relative amounts of intact HK and cHK were quantified using Image Studio Lite 5.2.
EV polyP detection
To characterize EV-associated polyP, EVs were preincubated with HBS, DNase I, RNase A, or CIP at 37°C for 1 hour, solubilized, and analyzed on a Tris-Borate-EDTA gel (EC62252; Thermo Fisher Scientific).33 The gel was stained with buffer (2.5% Ficoll-400, 10 mM EDTA, 3.3 mM Tris-HCl) containing 50 μg/mL DAPI and destained with the same buffer not containing DAPI. Long-, medium-, and short-chain polyP (gifts from James Morrissey37 ) were used to estimate the chain length of EV-associated polyP.
Effect of phosphatase treatment on ligand binding by EVs
EV were incubated with bacterial exopolyphosphatase (PPX; gift from Thomas Renné38 ; 200 µg/mL, 37°C, 1 hour), which degrades polyP, or with buffer alone. EVs were then incubated with 10 µg/mL an Alexa Fluor 488 (AF488)-labeled mutant PPX construct lacking catalytic domains 1 and 2 (PPXΔ12).38 EV-bound PPXΔ12-AF488 was measured in fluorescence mode with a 488-nm excitation laser using a ZetaView NTA PMX-120. Anti-human CD81-AF488 antibody (20 μg/mL, FAB4615G; R&D Systems) was used as a control ligand.
Similar studies were performed using AF488-labeled FXII. Conjugation of AF488 to PPXΔ12 and FXII was performed according to the manufacturer’s instructions (Alexa Fluor 488 Labeling Kit; Invitrogen).
In vivo thrombosis studies
To assess the role of the contact system in thrombosis induced by EVs we used EV prepared from L3.6 cells, some of which had been pretreated with CIP and others in buffer alone. Briefly, wild-type (WT), FXII-deficient (f12−/−), PK-deficient (klkb1−/−), or HK-deficient (kgn1−/−) mice were anesthetized and injected via the exposed inferior vena cava with 0.3 to 0.4 µg of EV per gram of body weight in 150 µL of PBS containing 0.6 µg epinephrine per gram of body weight.39 Following injections, animals were observed until death, at which time the lungs were exposed, the trachea was dissected, and both lungs were infused with 1 mL of OCT (Tissue-Tek, Torrance CA).40 One lung was fixed in 4% paraformaldehyde and the other was frozen in OCT and preserved at −80°C. Paraformaldehyde-fixed tissues were cut into 10-µm sections, stained with hematoxylin and eosin (H&E), scanned at 40× on a Leica SCN 400 Slide Scanner equipped with a Hamamatsu line sensor color camera, and examined using a digital microscope at ×10 magnification . The number of vessel occlusions per 10 random high-powered fields (100×) was determined from H&E staining. Deposition of platelets and fibrin in thrombosed vessels was assessed by immunofluorescence staining using a rat anti-mouse CD42c antibody (emfret ANALYTICS; cat. #M050-0) and a monoclonal antibody to murine fibrin (59D8; a gift from Hartmut Weiler41 ), followed by incubation with donkey anti-rat IgG–Alexa Fluor 594 or goat anti-mouse IgG–Alexa Fluor 488 (Invitrogen), respectively.
Capillary immunoassay of HK and cHK in plasma
HK and cHK in plasma were quantified using a Wes capillary immunoblotting system (ProteinSimple) and human monoclonal anti-HK (KNG17A12; Molecular Innovations). Standard curves were generated by adding known quantities of purified HK (Enzyme Research Laboratories) to HK-deficient plasma (Affinity Biologics). Standard curves for cHK were generated by adding known quantities of purified cHK (Enzyme Research Laboratories) to normal plasma deactivated with Laemmli buffer and boiled for 10 minutes at 95°C. Diluted plasma samples were mixed 4:1 with Fluorescent Master Mix and heated for 5 minutes at 95°C. Samples were loaded into a 24-well 12- to 230-kDa Wes detection plate and analyzed. Peak areas of immunoreactivity and standard curves were used to quantify HK and cHK concentrations.
FXII activation by EVs from patients with cancer
EVs from patients with cancer and normal healthy donors were isolated using a Pan-Exosome Isolation Kit, Pan (Miltenyi Biotec). Citrated human plasma (300 µL) was centrifuged at 15 000g at 4°C for 5 minutes, and the supernatant was incubated with pan-exosome beads conjugated with anti-human CD9/CD63/CD81. The beads were collected using a magnet. EV binding to beads was confirmed by flow cytometric analysis of the immunoprecipitated EV using FITC mouse anti-human CD9 or PE mouse anti-human CD63 (both from BD Biosciences).
To assess the ability of the immunoprecipitated EV to activate FXII, control beads or beads that had been incubated with plasma from healthy controls or patients with cancer were added to aliquots of the same normal plasma. FXII activation was assessed by hydrolysis of S-2302.
Statistics
Data are shown as means ± standard error of the mean (SEM). Statistical analyses were performed using GraphPad Prism 8.0 (GraphPad Software, San Diego, CA). Significance was determined using a 2-tailed unpaired Student t test or analysis of variance (ANOVA) with multiple comparisons test and P values < .05. Time-to-event data were modeled using Cox proportional hazards regression models with continuous predictors included as 2 degrees of freedom restricted cubic splines, which allow for nonlinear relationships between log hazards and predictors, as implemented in the cph and rcs functions of the R package rms.42 Significance of predictors was assessed using likelihood ratio tests, with P values < .05 as the threshold. Event times were censored at death times when the primary event did not include death. Violations of the proportional hazard assumptions were assessed using residuals plots (R function cox.zph).
Results
EV characterization
EV from all cell types eluted with a similar profile following size exclusion chromatography, with the highest particle numbers observed in fractions 7 to 10 (Figure 1A); plasma proteins were eluted in fractions 18 to 22 (Figure 1B). EV from L3.6 cells and HDFs that eluted in fractions 7 to 10 were analyzed for tetraspanin content by immunoblotting with antibodies to CD9, CD63, and CD 81 (Figure 1C). Tetraspanins were present in fractions 8 to 12, with peak expression in fractions 8 and 9. Nanoparticle tracking analysis of HDFs and L3.6 EV within fractions 7 to 10 revealed a mean particle size of 99.3 and 97.6 nm, respectively (Figure 1D). EVs exhibited the expected bilamellar appearance by electron microscopy (Figure 1E).

Characterization of EV. Particle numbers (A) and relative protein concentrations (B) in the fractions eluted following qEV size-exclusion chromatography. Data are mean ± SEM. (C) Tetraspanin (CD9, CD63, and CD81) content of purified EVs was analyzed in the indicated elution fractions by immunoblotting. (D) Size distribution of pooled fractions 7 to 10 from HDFs (left panel) and L3.6 cells (right panel). (E) Electron microscopy image of EV from HDFs (left panel) and L3.6 cells (right panel).
Characterization of EV. Particle numbers (A) and relative protein concentrations (B) in the fractions eluted following qEV size-exclusion chromatography. Data are mean ± SEM. (C) Tetraspanin (CD9, CD63, and CD81) content of purified EVs was analyzed in the indicated elution fractions by immunoblotting. (D) Size distribution of pooled fractions 7 to 10 from HDFs (left panel) and L3.6 cells (right panel). (E) Electron microscopy image of EV from HDFs (left panel) and L3.6 cells (right panel).
Cancer cell–derived EVs express polyP
We assessed polyP content of EV derived from the cell lines under study43 (Figure 2A) using a standard curve prepared with long-chain polyP (supplemental Figure 2A). EVs from L3.6 and H1975 cells expressed the greatest amount of polyP (9.35 ± 1.45 and 9.43 ± 1.17 ng/µg EV protein, respectively), with lesser amounts expressed by EV derived from HT29 and U937 cells (6.89 ± 0.59 and 6.11 ± 0.75 ng/µg, respectively). Cancer cell EVs expressed more polyP than did HDF EV (4.58 ± 0.43 ng/µg).
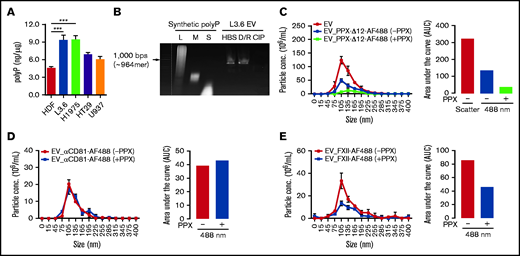
polyP on EV derived from cancer cells mediates ligand binding. (A) The amount of polyP associated with cancer cell–derived EV was estimated by comparison with a standard curve (supplemental Figure 2A). (B) Detection of polyP on L3.6-derived EVs following incubation with buffer (HBS), DNase I and RNase A (D/R), or CIP. polyP of different chain lengths was used to estimate the size of EV-associated polyP. L3.6-derived EVs were preincubated with buffer or E coli PPX, and the binding of PPX-Δ12–AF488 (C), anti-CD81–AF488 (D), and or FXII-AF488 (E) was measured. The number of total EV was determined in scatter mode, and the number of ligand-binding EV was determined in fluorescence mode using a 488-nm excitation laser. The differences in fluorescent particle numbers were compared by estimating the area under each curve (AUC) using GraphPad Prism 8. Bars represent means ± SEM. ***P < .001, 1-way ANOVA with multiple comparisons. L, long; M, medium; S, short.
polyP on EV derived from cancer cells mediates ligand binding. (A) The amount of polyP associated with cancer cell–derived EV was estimated by comparison with a standard curve (supplemental Figure 2A). (B) Detection of polyP on L3.6-derived EVs following incubation with buffer (HBS), DNase I and RNase A (D/R), or CIP. polyP of different chain lengths was used to estimate the size of EV-associated polyP. L3.6-derived EVs were preincubated with buffer or E coli PPX, and the binding of PPX-Δ12–AF488 (C), anti-CD81–AF488 (D), and or FXII-AF488 (E) was measured. The number of total EV was determined in scatter mode, and the number of ligand-binding EV was determined in fluorescence mode using a 488-nm excitation laser. The differences in fluorescent particle numbers were compared by estimating the area under each curve (AUC) using GraphPad Prism 8. Bars represent means ± SEM. ***P < .001, 1-way ANOVA with multiple comparisons. L, long; M, medium; S, short.
Using a nondenaturing Tris-Borate-EDTA gel, we determined that cancer cell–derived EVs expressed long-chain polyP, with a chain length of ∼960 phosphate monomers.33 PolyP was removed from EV by treatment with CIP,44 as well as Escherichia coli PPX (data not shown); DNase and RNase did not eliminate polyP expression (Figure 2B). Electron microscopy studies demonstrated that EV remained intact following CIP treatment (supplemental Figure 2B).
We examined the ability of polyP to mediate ligand binding to EV, initially using PPX-Δ12.38 Binding of PPX-Δ12–AF488 to EV before and after treatment with PPX was calculated by comparing the difference in fluorescent particle number, as determined by the measurement of the area under each binding curve. We observed that not all EV bound PPX-Δ12–AF488, suggesting heterogeneous polyP expression (Figure 2C). Pretreatment of EVs with PPX reduced the binding of PPX-Δ12–AF488 by 73%, demonstrating a key role for polyP in binding (Figure 2C). We did not observe any changes in the binding of AF488-labeled anti-CD81 antibody to EV after PPX treatment (Figure 2D).
Cancer cell–derived EVs activate FXII in human plasma
Using a chromogenic assay based on hydrolysis of S-2302, we initially observed that EV from L3.6, H1975, and HT29 cells induced FXII activation in NHP in a time-dependent manner (Figure 3A). In subsequent studies, the relative amounts of FXIIa generated was estimated by comparison with a standard curve prepared by adding known amounts of FXIIa to NHP (supplemental Figure 3). The relative amount of FXII activation caused by EV was dependent on EV concentration, with little activation caused by HDFs or U937 EVs (Figure 3B). The ability of EVs to activate FXII was inhibited by pretreatment with CIP, but not with DNase or RNase (Figure 3C).
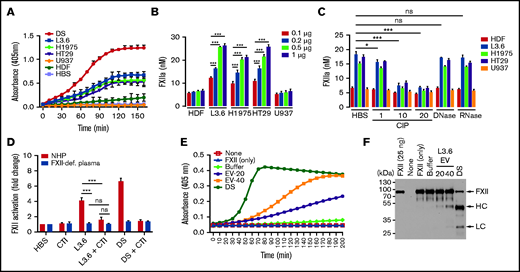
FXII activation by EVs derived from cancer cell lines. (A) Activation of FXII induced by EV from cancer cells (L3.6, H1975, HT29, and U937) and HDFs. (B) FXIIa generation by cancer cell–derived EV. EV-induced S-2302 hydrolysis was estimated at 90 minutes after addition of the indicated amount of EV (determined by protein concentration) and comparison with a FXIIa standard curve (supplemental Figure 3). (C) FXIIa generated by cancer cell–derived EV treated with DNase I (10 U/mL), RNase A (10 U/mL), and CIP (1, 10, and 20 U/mL). (D) Effect of CTI on FXII activation induced by L3.6 EV in NHP and FXII-deficient (FXII-def.) plasma. Hydrolysis of S-2302 in plasma incubated with EV in the presence or absence of CTI was compared. (E) Activation of purified FXII (375 nM) by EV. EVs (20 or 40 µg protein) were incubated in the presence of FXII (375 nM) and S-2302 and A405 were monitored for 90 minutes. (F) FXII cleavage in the presence of L3.6 EV was assessed by immunoblotting using the reaction mixture in (D). The FXII heavy chain (HC) and light chain (LC) were detected under reducing conditions using goat anti-human FXII polyclonal antibody. Bars represent means ± SEM. *P < .05, **P < .01, ***P < .001, 2-way ANOVA with multiple comparisons. ns, not significant.
FXII activation by EVs derived from cancer cell lines. (A) Activation of FXII induced by EV from cancer cells (L3.6, H1975, HT29, and U937) and HDFs. (B) FXIIa generation by cancer cell–derived EV. EV-induced S-2302 hydrolysis was estimated at 90 minutes after addition of the indicated amount of EV (determined by protein concentration) and comparison with a FXIIa standard curve (supplemental Figure 3). (C) FXIIa generated by cancer cell–derived EV treated with DNase I (10 U/mL), RNase A (10 U/mL), and CIP (1, 10, and 20 U/mL). (D) Effect of CTI on FXII activation induced by L3.6 EV in NHP and FXII-deficient (FXII-def.) plasma. Hydrolysis of S-2302 in plasma incubated with EV in the presence or absence of CTI was compared. (E) Activation of purified FXII (375 nM) by EV. EVs (20 or 40 µg protein) were incubated in the presence of FXII (375 nM) and S-2302 and A405 were monitored for 90 minutes. (F) FXII cleavage in the presence of L3.6 EV was assessed by immunoblotting using the reaction mixture in (D). The FXII heavy chain (HC) and light chain (LC) were detected under reducing conditions using goat anti-human FXII polyclonal antibody. Bars represent means ± SEM. *P < .05, **P < .01, ***P < .001, 2-way ANOVA with multiple comparisons. ns, not significant.
To further demonstrate the activation of FXII by EV, we assessed the ability of L3.6 EVs to cause hydrolysis of S-2302 in FXII-deficient plasma, as well as in the presence of CTI, a FXIIa inhibitor (Figure 3D). As expected, FXII activation by L3.6 EV was not detectable in FXII-deficient plasma. CTI also significantly reduced FXII activation in NHP.
To determine whether plasma cofactors were required for activation of FXII by EV, we assessed the ability of L3.6 EVs to activate purified FXII. Incubation of EVs with FXII enhanced FXIIa generation, as measured by S-2302 hydrolysis (Figure 3E) and cleavage of FXII (Figure 3F). Heating EV to 100°C to destroy protease activity did not block their ability to activate FXII (supplemental Figure 4).
EV-induced activation of FXII correlates with cleavage of HK
Because HK cleavage is a marker of contact activation,27-29 we used immunoblotting to determine whether EV caused cleavage of HK in parallel with S-2302 hydrolysis. EV from L3.6 and H1975 cells caused generation of cHK in NHP in a concentration-dependent manner (Figure 4A); using LI-COR Biosciences–based quantification, we determined that the ratio of cHK/HK increased from 8% to 16%, from 6% to 17%, and from 5% to 11% when EVs from L3.6, H1975, and HT29 cells, respectively, were used at concentrations of 0.5 and 1 μg/mL (Figure 4B). EV from HDFs or U937 cells caused minimal cleavage of HK.
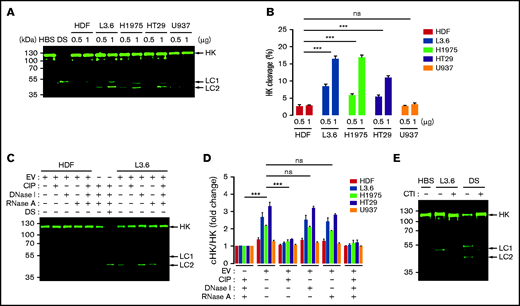
Cleavage of HK caused by cancer cell–derived EV. (A) HK cleavage in NHP incubated with cancer cell–derived EV (0.5 and 1 μg/mL) and analyzed by immunoblotting. (B) HK cleavage by EVs from (A) expressed as the percentage of cHK relative to intact HK. (C) HK cleavage 60 minutes after the addition of L3.6 EV following treatment of EV with CIP, DNase I, or RNase A. DS was used as a positive control. (D) Effect of CIP, DNase I, and RNase A on the cleavage of HK by EV from cancer cells and HDFs; the ratio of cHK/HK was determined by immunoblotting and infrared quantification. (E) Effect of CTI on L3.6 EV-induced cHK generation. L3.6 EV were incubated with NHP in the presence or absence of CTI (10 μg/mL) for 60 minutes. For HK cleavage analysis, a polyclonal rabbit anti-human HK antibody (D5; supplemental Figure 1) was used to detect HK and cHK. Bars represent means ± SEM. ***P < .001, 2-way ANOVA with multiple comparisons. LC1, light chain 1 (54 kDa); LC2, light chain 2 (47 kDa). ns, not significant.
Cleavage of HK caused by cancer cell–derived EV. (A) HK cleavage in NHP incubated with cancer cell–derived EV (0.5 and 1 μg/mL) and analyzed by immunoblotting. (B) HK cleavage by EVs from (A) expressed as the percentage of cHK relative to intact HK. (C) HK cleavage 60 minutes after the addition of L3.6 EV following treatment of EV with CIP, DNase I, or RNase A. DS was used as a positive control. (D) Effect of CIP, DNase I, and RNase A on the cleavage of HK by EV from cancer cells and HDFs; the ratio of cHK/HK was determined by immunoblotting and infrared quantification. (E) Effect of CTI on L3.6 EV-induced cHK generation. L3.6 EV were incubated with NHP in the presence or absence of CTI (10 μg/mL) for 60 minutes. For HK cleavage analysis, a polyclonal rabbit anti-human HK antibody (D5; supplemental Figure 1) was used to detect HK and cHK. Bars represent means ± SEM. ***P < .001, 2-way ANOVA with multiple comparisons. LC1, light chain 1 (54 kDa); LC2, light chain 2 (47 kDa). ns, not significant.
Pretreatment of EVs with CIP, but not DNase I or RNase A, inhibited EV-induced cHK generation (Figure 4C-D). Likewise, CTI inhibited the ability of L3.6 EVs to generate cHK (Figure 4E).
EVs from L3.6 pancreatic cancer cells induce pulmonary emboli through a contact activation–dependent pathway
EVs derived from L3.6 cells were injected into the inferior vena cava (IVC) of WT, f12−/−, kng1−/−, or klkb1−/− mice. Pulmonary emboli developed in all WT mice (Figure 5A), with thrombi demonstrating extensive platelet and fibrin deposition (Figure 5B-C). However, the extent of thrombi varied among different strains, with the largest thrombotic burden observed in WT mice, the lowest burden in f12−/− mice, and intermediate amounts in kng1−/− and klkb1−/− mice (Figure 5D). The thrombus burden was reduced significantly compared with WT mice in all of the contact factor–deficient strains.
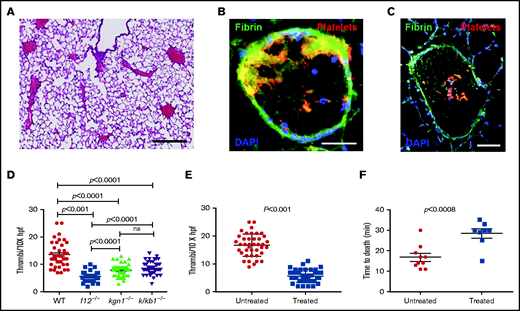
Pulmonary emboli in mice treated with EV derived from L3.6 cells. (A) Paraffin section of lung from a WT mouse after L3.6 EV infusion into the IVC (original magnification ×5; H&E stain). Scale bar, 500 μm. Frozen sections of lung following EV infusion into the IVC and staining for platelet GP1b (CD42c, red) or fibrin (monoclonal antibody 59D8, green) at original magnification 320 (scale bar, 100 mm). (B) At original magnification 310 (scale bar, 200 mm. (C) DAPI (blue) stains cell nuclei. (D) Number of thrombi observed in a random 10× field following infusion of L3.6 EVs into WT C57BL/6, f12−/−, kng1−/−, and klkb1−/− mice. (E) Effect of preincubation of L3.6 EV with buffer alone (untreated) or CIP (treated) prior to IVC infusion on density of lung thrombi, measured per ×10× field. (F) Effect of preincubation of L3.6 EV with buffer alone (untreated) or CIP (treated) on time to death after IVC infusion. Each point represents an individual animal. Bars in graphs depict 95% confidence intervals , and comparisons between groups were performed using ANOVA and Student t tests. hpf, high-powered field; ns, not significant.
Pulmonary emboli in mice treated with EV derived from L3.6 cells. (A) Paraffin section of lung from a WT mouse after L3.6 EV infusion into the IVC (original magnification ×5; H&E stain). Scale bar, 500 μm. Frozen sections of lung following EV infusion into the IVC and staining for platelet GP1b (CD42c, red) or fibrin (monoclonal antibody 59D8, green) at original magnification 320 (scale bar, 100 mm). (B) At original magnification 310 (scale bar, 200 mm. (C) DAPI (blue) stains cell nuclei. (D) Number of thrombi observed in a random 10× field following infusion of L3.6 EVs into WT C57BL/6, f12−/−, kng1−/−, and klkb1−/− mice. (E) Effect of preincubation of L3.6 EV with buffer alone (untreated) or CIP (treated) prior to IVC infusion on density of lung thrombi, measured per ×10× field. (F) Effect of preincubation of L3.6 EV with buffer alone (untreated) or CIP (treated) on time to death after IVC infusion. Each point represents an individual animal. Bars in graphs depict 95% confidence intervals , and comparisons between groups were performed using ANOVA and Student t tests. hpf, high-powered field; ns, not significant.
We also treated L3.6 EVs with CIP prior to infusion into WT mice. Compared with PBS-treated EV, the number of pulmonary emboli in mice that received phosphatase-treated EVs was significantly reduced (Figure 5E). These mice also demonstrated a prolonged time to death (28 ± 6 minutes) compared with mice that received untreated EV (16.8 ± 1.8 minutes; P < .008) (Figure 5F).
Elevated levels of cHK in patients with cancer
To examine the clinical relevance of our findings, we measured levels of cHK in patients with cancer. Using immunoblotting, analysis of plasma from 5 patients with pancreatic cancer demonstrated increased cHK, which was detected using antibodies to HK domain 5 (Figure 6A) or an HK heavy chain–specific antibody (Figure 6B).
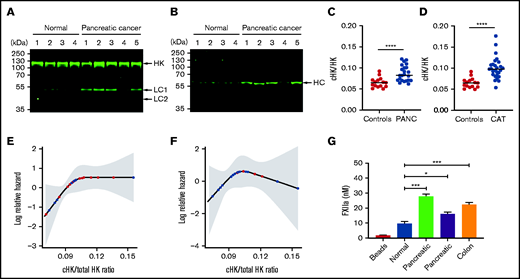
Increased levels of cHK in plasma from patients with cancer. (A) HK cleavage in plasma from patients with pancreatic cancer and healthy donors (normal) was determined by immunoblotting using antibody to HK domain 5. (B) The same plasmas as in (A) were analyzed using an antibody specific for the C terminus of the free HK heavy chain (HC) after reduction (HKa1; supplemental Figure 1). (C) Ratio of cHK/HK in plasma from healthy individuals and patients with pancreatic cancer (PANC) (n = 26), determined by Wes capillary immunoblotting. (D) Ratio of cHK/HK in healthy individuals and patients presenting to the cancer thrombosis clinic (n = 21) with symptoms of VTE, as determined by Wes capillary immunoblotting. Bars represent means ± SEM. ***P < .001, ****P < .0001, unpaired Student t test with Welch’s correction. (E) Log relative hazard of VTE or death vs cHK/total HK ratio from the Cox proportional hazard modeling of patients with different types of cancer. Each red dot indicates a VTE or death event, and each black dot indicates a censored event; the shaded area is the 95% confidence interval for the log hazard. (F) Log relative hazard of VTE only (censored at death) vs cHK/total HK ratio from the Cox proportional hazard modeling of patients with different types of cancer. Each red dot indicates a VTE event, and each black dot indicates a censored event; the shaded area is the 95% confidence interval for the log hazard. (G) FXII activation by EVs immunopurified from a healthy donor or from patients with pancreatic or colon cancer. Data are means ± SEM. *P < .05, ***P < .001, 1-way ANOVA with a unpaired Student t test with multiple comparisons.
Increased levels of cHK in plasma from patients with cancer. (A) HK cleavage in plasma from patients with pancreatic cancer and healthy donors (normal) was determined by immunoblotting using antibody to HK domain 5. (B) The same plasmas as in (A) were analyzed using an antibody specific for the C terminus of the free HK heavy chain (HC) after reduction (HKa1; supplemental Figure 1). (C) Ratio of cHK/HK in plasma from healthy individuals and patients with pancreatic cancer (PANC) (n = 26), determined by Wes capillary immunoblotting. (D) Ratio of cHK/HK in healthy individuals and patients presenting to the cancer thrombosis clinic (n = 21) with symptoms of VTE, as determined by Wes capillary immunoblotting. Bars represent means ± SEM. ***P < .001, ****P < .0001, unpaired Student t test with Welch’s correction. (E) Log relative hazard of VTE or death vs cHK/total HK ratio from the Cox proportional hazard modeling of patients with different types of cancer. Each red dot indicates a VTE or death event, and each black dot indicates a censored event; the shaded area is the 95% confidence interval for the log hazard. (F) Log relative hazard of VTE only (censored at death) vs cHK/total HK ratio from the Cox proportional hazard modeling of patients with different types of cancer. Each red dot indicates a VTE event, and each black dot indicates a censored event; the shaded area is the 95% confidence interval for the log hazard. (G) FXII activation by EVs immunopurified from a healthy donor or from patients with pancreatic or colon cancer. Data are means ± SEM. *P < .05, ***P < .001, 1-way ANOVA with a unpaired Student t test with multiple comparisons.
We next analyzed a larger sample of patients with pancreatic cancer using quantitative capillary immunoblotting. Samples were obtained before the advent of any thrombotic events. This analysis confirmed the presence of elevated levels of cHK in patients with pancreatic cancer (Figure 6C; P < .0001). However, only 6 of the 23 patients in this cohort developed thrombosis; given the small sample size, we were unable to demonstrate a statistically significant increase in cHK in these patients.
We used the same approach to analyze plasma obtained from patients presenting to our cancer thrombosis clinic with signs and/or symptoms of venous thromboembolism (VTE) (Figure 6D). We observed higher levels of cHK in patients with cancer than in normal controls (P < .0001). However, again because of the small sample size that included only 6 patients with thrombosis, we could not demonstrate that cHK/total HK differentiated patients with VTE from those without.
Finally, we prospectively analyzed the cohort of 21 patients seen in the cancer thrombosis clinic, who did not have thrombosis at the time of presentation, for the subsequent development of VTE. The log relative hazard for death and/or developing VTE, or just for developing VTE, as a function of the cHK/total HK ratio is depicted in Figure 6E and 6 F, respectively. Because of the small sample sizes (13/21 for VTE or death and 6/21 for VTE only), these analyses were unable to demonstrate a statistically significant increased risk for future events in patients with elevated cHK/HK (P = .11 and 0.39, respectively), although a clear trend toward increased risk is seen with cHK values in the range from 0.075 to 0.105.
Although these studies demonstrate activation of the contact system in cancer, they do not prove that circulating EVcontribute. To address this question, we used beads coated with antibodies to CD81, CD63, and CD9 to immunoprecipitate EVs from a healthy individual and 3 patients (2 with pancreatic cancer and 1 with colorectal cancer). Next, the ability of immunoprecipitated EVs to activate FXII was assessed. As shown in Figure 6G, a small amount of FXIIa was generated by EVs from the healthy donor, but greater activation was induced by EVs from each of the patients with cancer.
Discussion
Our findings demonstrate that long-chain polyP is expressed on EV derived from cancer and nontransformed cells. On the EV surface, polyP mediates binding of FXII. As a consequence of FXII binding, EV activate FXII to FXIIa, leading to activation of the contact system and intrinsic coagulation pathway.
Thrombosis is a common complication in patients with cancer.12,13 Multiple mechanisms for the development of thrombosis have been proposed, with significant interest in the role of tissue factor–expressing EVs.18-20,45 Levels of tissue factor–positive EV correlate with the development of thrombosis in patients with pancreatic cancer,22,46 although such a relationship has not been demonstrated for other cancer types.22,23,47-49 There has been comparatively little study of the intrinsic FXII-mediated pathway in cancer. Our studies demonstrate that activation of this pathway occurs commonly in patients with cancer (Figure 6A-D). Moreover, the ability of EV from different types of cancer cells to cause FXII activation varies and correlates with the strength of the association of the parental cancer cells with thrombosis.50 These findings suggest that cHK may serve as a biomarker of thrombosis in cancer, although addressing this question will require larger studies.
polyP-expressing EV induced pulmonary emboli in mice, whereas pretreatment of EV with phosphatase impaired their prothrombotic activity. Moreover, the ability of polyP-expressing EVs to induce pulmonary emboli was reduced significantly in mice deficient in FXII, prekallikrein, or HK; these findings implicate a critical role for the contact activation system in EV-induced thrombosis. In contrast to the study by Nickel et al,33 we found that klkb1−/− mice were also protected from the development of EV-induced thrombi in vivo; the reasons for this discrepancy will require additional investigation.
There is little information available concerning the role of polyP in cancer cells, the mechanisms by which polyP becomes associated with cancer cell–derived EVs, and why only a subpopulation of EVs express polyP. Although the biosynthetic pathway of polyP in mammalian cells is not well understood, the cellular content of polyP may depend upon the activity of inositol hexakisphosphate kinase 1 (IP6K1).51 Mammalian cells lacking IP6K1 exhibit impaired dynein-dependent trafficking, including endosomal sorting and vesicle movement,52 which may lead to decreased exosome release53 ; genetic ablation of Ip6k1 in mice results in reduced platelet polyP.54 Data from the Human Protein Atlas suggest higher expression of IP6K1 in colorectal, pancreatic, and prostate33 cancers; these findings may account, in part, for the elevated levels of polyP on pancreatic cancer cell–derived EV, as well as by prostasomes.33
Our study has several limitations, the most important of which is that we focused on polyP and contact activation but did not address other potential prothrombotic mechanisms of cancer cell EVs, such as tissue factor. However, 1 study demonstrated that prostasomes may enhance tissue factor and intrinsic pathway–mediated coagulation activation,33,39 and this is likely the case with the EVs used in this study, as well; indeed, tissue factor antigen expression by L3.6 cells, but not the other cell lines, was detected by immunoblotting (supplemental Figure 5). Nevertheless, our clinical studies demonstrate activation of the contact system in a high proportion of patients with cancer and confirm that patient-derived EV activate FXII in plasma. Although our clinical cohort lacks the power to demonstrate that cHK is a biomarker for thrombosis, this limitation will be addressed in future studies.
In summary, our studies suggest that EV from patients with cancer, as well as cancer cells, express increased amounts of polyP compared with healthy individuals or nontransformed cell lines and that these polyP-expressing EVs bind FXII and activate the contact system. These EVs induce thrombosis in animal models and are less effective in doing so after treatment with phosphatase or in mice lacking key proteins of the contact system. These findings suggest a role for polyP-expressing EV in cancer-associated thrombosis and raise the possibility that polyP inhibitors55 prevent thrombosis in patients with cancer without increasing bleeding risk.
Acknowledgments
The authors thank James H. Morrissey (University of Michigan, Ann Arbor, MI) for the kind gift of polyP, Thomas Renné (University Medical Center-Hamburg, Germany) for providing expression constructs for PPX and PPXΔ12, and Hartmut Weiler (Blood Research Institute, Milwaukee, WI) for providing monoclonal antibody 59D8.
This work was supported by grant U01 HL143402 from the National Institutes of Health National Heart, Lung, and Blood Institute Consortium Linking Oncology with Thrombosis (K.R.M. and A.A.K.), as well as by a grant from the Cleveland Clinic VeloSano foundation. A.A.K. is supported by the Sondra and Stephen Hardis Chair in Oncology Research.
Authorship
Contribution: Y.J.S. and V.C. designed and performed experiments; S.S. and R.K.A. assisted with fluorescent ligand binding experiments; S.K. assisted with cell culture; D.Y. performed experiments; S.W. and E.P.F. designed and conducted ProteinSimple assays for HK and cHK and edited the manuscript; J.B. performed biostatistical analyses; A.S. and A.M. designed and analyzed the murine pulmonary emboli studies and edited the manuscript; A.A.K. provided clinical samples, designed experiments, and edited the manuscript; D.A. provided clinical samples and K.R.M. developed concepts, organized data, and wrote and edited the manuscript.
Conflict-of-interest disclosure: A.A.K. has received personal fees and nonfinancial support from Janssen, Bayer, Pfizer, Seattle Genetics, and Bristol-Myers Squibb; personal fees from Parexel, Pharmacyclics, Pharmacyte, and Medscape; and grants to his institution from Merck, Array, Bristol Myers Squibb, Leap, and Seattle Genetics, all outside of the submitted work. The remaining authors declare no competing financial interests.
Correspondence: Keith R. McCrae, Departments of Hematology/Oncology and Cardiovascular and Metabolic Sciences, 9500 Euclid Ave, CA6, Cleveland Clinic, Cleveland, OH 44195; e-mail: mccraek@ccf.org.

