

Key Points
Rapid donor cell engraftment with ex vivo–expanded, partially mismatched UCB grafts led to a reduction in sickle cell disease severity.
Strategies to minimize the risk of severe GVHD and infections are needed.

Abstract
Many patients with sickle cell disease (SCD) do not have HLA-matched related donors for hematopoietic stem cell transplantation (HSCT). Unrelated cord blood (UCB) is an alternative graft option but is historically associated with high graft failure rates, with inadequate cell dose a major limitation. Omidubicel is a nicotinamide-based, ex vivo–expanded UCB product associated with rapid engraftment in adults with hematologic malignancies. We hypothesized that increasing the UCB cell dose with this strategy would lead to improved engraftment in pediatric patients undergoing myeloablative HSCT for SCD. We report the outcomes of a phase 1/2 study in 13 patients with severe SCD who received omidubicel in combination with an unmanipulated UCB graft and 3 who received a single omidubicel graft. Grafts were minimally matched with patients at 4 of 6 HLA alleles. Median age at transplant was 13 years. A median CD34+ expansion of ∼80-fold was observed in omidubicel and led to rapid neutrophil engraftment (median, 7 days). Long-term engraftment was derived from the unmanipulated graft in most of the double cord blood recipients. Two of the 3 single omidubicel recipients also had sustained engraftment. Incidence of acute graft-versus-host disease (GVHD) was high, but resolved in all surviving patients. Event-free survival in the double cord group was 85% (median follow-up 4 years). All 3 patients in the single cord group were alive at 1 year after transplantation. Ex vivo expansion of UCB with omidubicel supports engraftment in patients with SCD. This approach to decreasing the incidence of GVHD should be optimized for general use in patients with SCD. This study was registered at www.clinicaltrials.gov as #NCT01590628.
Introduction
Sickle cell disease (SCD) is a common inherited disorder characterized by hemolytic anemia and vasculopathy leading to significant chronic morbidity and early mortality. Approximately 300 000 infants with SCD are born each year worldwide, predominantly in sub-Saharan Africa and India and increasingly in Europe and the United States,1 where the prevalence is ∼100 000.2 Allogeneic hematopoietic stem cell transplantation (HSCT) from an HLA-identical, related donor is the only widely accepted curative therapy for patients with SCD. It is generally reserved for those with severe disease and is curative in 90% to 95% of patients.3 However, most patients do not have an HLA-identical donor.4,5 Main alternatives considered for such patients are haploidentical and unrelated umbilical cord blood (UCB) donors.6,7
Unrelated UCB, whether HLA-matched or partially mismatched, is an attractive alternative donor option for HSCT, given its availability and tolerability of some HLA mismatching.7,8 Since the first related UCB transplant (UCBT) in 1988 and unrelated UCBT in 1993, >40 000 transplants have been performed, mostly unrelated, in adults and children for both malignant and nonmalignant diseases, demonstrating its long-term curative potential.7
Early reports of UCBT in patients with hemoglobinopathies raised concerns about an increased risk of graft failure.9,10 A higher cell dose threshold was identified as an important factor that influenced the rates of engraftment in this alloimmunized patient population. One of the emerging approaches to increasing the number of stem and progenitor cells without accelerating their differentiation is ex vivo expansion of the UCB graft.11-17 A pilot study of transplantation with omidubicel, an ex vivo–expanded UCB graft, in combination with an unmanipulated UCB graft in adults with hematologic malignancies (HMs), has demonstrated robust neutrophil engraftment, as well as decreased hospitalizations and infections. High, rapid engraftment rates were demonstrated in subsequent studies of omidubicel used as a stand-alone graft.18-21
We hypothesized that transplantation with omidubicel would lead to improved engraftment in patients with hemoglobinopathies who have undergone UCBT. We report the outcomes of a pilot study testing omidubicel, with and without an unmanipulated UCB graft, in patients with symptomatic SCD.
Methods
Study design
This was a phase 1/2, open-label, single-arm study in patients with SCD. In the double cord blood transplantation (DC) cohort, the safety and efficacy of omidubicel in combination with unmanipulated UCB was assessed in 13 patients who underwent HSCT from October 2012 through January 2018 at Duke University Medical Center (n = 12) and Cohen Children’s Medical Center (n = 1). The median duration of follow-up of surviving patients was 4 years (range, 2.3-7.5 years). Three patients received a transplant of single-unit omidubicel in the single cord blood transplantation (SC) cohort at Duke University Medical Center from May 2018 through September 2018. The data include 1 year of follow-up.
The study was approved by the institutional review boards of the participating institutions and national regulatory authorities and was performed in accordance with the International Conference on Harmonization Guidelines and Good Clinical Practice. All patients and/or legal guardians provided written informed consent before treatment.
Patient eligibility
Eligible patients were 2 to 45 years of age with severe SCD defined by the presence of at least 1 of the following: recurrent vaso-occlusive painful crises or acute chest syndrome, despite adequate supportive care measures (at least 3 events in the 2 years before enrollment); a neurologic event with deficits lasting more than 24 hours; abnormal cerebral imaging; or abnormal transcranial Doppler velocities on 2 separate occasions at least 1 month apart. Patients with 8 of 8 HLA-matched related or available unrelated donors were excluded. Adequate organ function and performance score ≥0 (Lansky or Karnofsky) were required. A hemoglobin S (HbS) level of <45%, achieved via transfusion or erythrocyte exchange, was required. The patients were required to have an autologous or unrelated graft source as a backup in the event of graft failure.
Conditioning regimen
The patients received myeloablative conditioning with either regimen A (hydroxyurea, busulfan, cyclophosphamide and horse ATG [eATG]), or regimen B (hydroxyurea, fludarabine, busulfan, and cyclophosphamide) (Table 1). The first 3 patients in the DC cohort received regimen A. The subsequent 10 patients in the DC cohort and first 2 patients in the SC cohort received regimen B. Patient SC-3 received regimen B with the addition of eATG.
Demographic and baseline characteristics
| DC | SC | |||
|---|---|---|---|---|
| Characteristic | n | % | n | % |
| Total | 13 | 100.0 | 3 | 100 |
| Sex, male | 4 | 30.8 | 2 | 66 |
| Age at enrollment, y | ||||
| 2-11 | 6 | 46.2 | 0 | 0 |
| 12-17 | 7 | 53.8 | 3 | 100 |
| Primary race, Black | 13 | 100.0 | 3 | 100 |
| Sickle cell or thalassemia disease type | — | — | ||
| HbSS | 13 | 100.0 | 2 | 66 |
| HbSS/β0 thalassemia | — | — | 1 | 33 |
| CMV seropositive | 7 | 53.8 | 2 | 66 |
| Performance status | ||||
| 100 | 6 | 46.2 | 2 | 66 |
| 90 | 3 | 23.1 | 0 | 0 |
| 80 | 4 | 30.8 | 0 | 0 |
| 70 | 0 | — | 1 | 33 |
| Three or more painful events in the past 2 y | 6 | 46.2 | 2 | 66 |
| ACS (≥2 episodes in the past 2 y) | 2 | 15.4 | 1 | 33 |
| Neurologic event | 4 | 30.8 | 1 | 33 |
| Abnormal MRI/MRA results | 5 | 38.5 | 1 | 33 |
| Long-term PRBC transfusions | 8 | 61.5 | 1 | 33 |
| Hydroxyurea use before enrollment | 10 | 76.9 | 2 | 66 |
| Conditioning regimen A | 3 | 23.0 | — | — |
| (−35 to −10 hydroxyurea, 30 mg/kg per d orally; −9 to −6 busulfan 1 mg/kg per dose, IV every 6 h × 16 doses; −5 to −2 cyclophosphamide 50 mg/kg per d × 4 doses; and −3 to −1 eATG 30 mg/kg per d × 3 doses) | ||||
| Conditioning regimen B * | 10 | 77.0 | 2 | 66 |
| (−35 to −15 hydroxyurea 30 mg/kg/day orally; −14 to −10 fludarabine 35 mg/m2 IV daily × 5 days; −9 to −6 busulfan 1 mg/kg/dose IV every 6 h × 16 doses; and −5 to −2 cyclophosphamide 50 mg/kg per day × 4 doses) | ||||
| Conditioning regimen B (modified)* | — | — | 1 | — |
| (−12 to −10 eATG 30 mg/kg per day) | ||||
| DC | SC | |||
|---|---|---|---|---|
| Characteristic | n | % | n | % |
| Total | 13 | 100.0 | 3 | 100 |
| Sex, male | 4 | 30.8 | 2 | 66 |
| Age at enrollment, y | ||||
| 2-11 | 6 | 46.2 | 0 | 0 |
| 12-17 | 7 | 53.8 | 3 | 100 |
| Primary race, Black | 13 | 100.0 | 3 | 100 |
| Sickle cell or thalassemia disease type | — | — | ||
| HbSS | 13 | 100.0 | 2 | 66 |
| HbSS/β0 thalassemia | — | — | 1 | 33 |
| CMV seropositive | 7 | 53.8 | 2 | 66 |
| Performance status | ||||
| 100 | 6 | 46.2 | 2 | 66 |
| 90 | 3 | 23.1 | 0 | 0 |
| 80 | 4 | 30.8 | 0 | 0 |
| 70 | 0 | — | 1 | 33 |
| Three or more painful events in the past 2 y | 6 | 46.2 | 2 | 66 |
| ACS (≥2 episodes in the past 2 y) | 2 | 15.4 | 1 | 33 |
| Neurologic event | 4 | 30.8 | 1 | 33 |
| Abnormal MRI/MRA results | 5 | 38.5 | 1 | 33 |
| Long-term PRBC transfusions | 8 | 61.5 | 1 | 33 |
| Hydroxyurea use before enrollment | 10 | 76.9 | 2 | 66 |
| Conditioning regimen A | 3 | 23.0 | — | — |
| (−35 to −10 hydroxyurea, 30 mg/kg per d orally; −9 to −6 busulfan 1 mg/kg per dose, IV every 6 h × 16 doses; −5 to −2 cyclophosphamide 50 mg/kg per d × 4 doses; and −3 to −1 eATG 30 mg/kg per d × 3 doses) | ||||
| Conditioning regimen B * | 10 | 77.0 | 2 | 66 |
| (−35 to −15 hydroxyurea 30 mg/kg/day orally; −14 to −10 fludarabine 35 mg/m2 IV daily × 5 days; −9 to −6 busulfan 1 mg/kg/dose IV every 6 h × 16 doses; and −5 to −2 cyclophosphamide 50 mg/kg per day × 4 doses) | ||||
| Conditioning regimen B (modified)* | — | — | 1 | — |
| (−12 to −10 eATG 30 mg/kg per day) | ||||
ACS, acute chest syndrome; MRA, magnetic resonance angiography; MRI, magnetic resonance imaging; PRBC, packed red blood cell; TCD, transcranial Doppler ultrasound.
The last patient in the SC cohort received additional eATG 30 mg/kg per day on days −12 to −10.
Hydroxyurea was administered at 30 mg/kg per day orally on days −35 to −15. Busulfan was initiated at 1 mg/kg IV every 6 hours, on days −9 to −6, for 16 doses. Busulfan pharmacokinetics were assessed at the first dose, and adjustments were made to achieve a steady-state concentration of 600 to 900 ng/mL, corresponding to myeloablative exposure (area under the curve, 60-85 mg/L per hour). The cyclophosphamide dose was 50 mg/kg per day IV, on days −5 to −2, for 4 doses. The eATG dose was 30 mg/kg, on days −3 to −1 in regimen A and days −12 to −10 for patient SC-3 in regimen B. The fludarabine dose was 35 mg/m2 on days −14 to −10. Graft-versus-host disease (GVHD) prophylaxis with cyclosporine and mycophenolate was started on day −3 and continued for at least 180 and 45 days, respectively.
Graft selection
Patients had 2 (DC) or 1 (SC) unrelated donor cord blood units (CBUs) identified. The units were minimally HLA matched with recipients at 4 of 6 to 6 of 6 HLA loci class 1 (HLA-A, B at low resolution) and class 2 (HLA-DRB1 at high resolution) and, in the DC cohort, with each other at 3 of 6 to 6 of 6 HLA loci. Double mismatch at any locus (A, B, or DRB1) was not permitted. The CBU was excluded if the patient had donor-specific HLA antibodies directed against any allele. The CBU designated for omidubicel production contained a precryopreserved (postprocessing) total nucleated cell (TNC) count of ≥1.8 ×109 cells, a dose of ≥1.8 × 107 cells per kg body weight, and a total CD34+ cell count of ≥8 × 106 or ≥7 × 106 cells for patients weighing ≥25 or 25 kg, respectively. The unmanipulated CBU used in the DC cohort contained a precryopreserved (postprocessing) TNC dose of ≥3.5 × 107/kg. CBUs were required to be volume reduced and red blood cell depleted before cryopreservation and were obtained from public cord blood banks that met national regulatory standards.
Omidubicel production
The UCB unit selected for omidubicel production underwent CD133 selection. The CD133+ fraction, referred to as the cultured fraction (CF), was expanded ex vivo for 21 days in the presence of early acting stem cell cytokines and nicotinamide.21 The CF, consisting of expanded early hematopoietic and myeloid progenitor cells, was shipped fresh to the transplant center early in the trial (n = 5); subsequently, the CF was cryopreserved before shipping (n = 11). The CD133− fraction, designated as the noncultured fraction (NF), consisted of mature lymphoid and myeloid cells. The NF was cryopreserved and sent to the transplant center where it was stored until the day of transplant. For DC patients on transplant day, the unmanipulated CBU was infused first, followed by sequential infusion of omidubicel CF and NF a minimum of 4 hours later. CF and NF were infused into the SC patients sequentially. The CD34+ and CD3+ cell content of the graft was quantified before cryopreservation of the product.
End points and statistical methods
The primary end points were infusion toxicity, defined as grade 4 or 5 toxicity within 24 hours of omidubicel infusion, and neutrophil engraftment, defined as absolute neutrophil count (ANC) of ≥0.5 × 103/μL on 3 consecutive measurements on different days. Additional end points included platelet engraftment (≥50 × 103/μL without transfusion), acute and chronic GVHD, infections, adverse events graded by National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03 (https://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm), transplant-related mortality (TRM), event-free survival (EFS), and overall survival (OS).
In the DC cohort, cumulative incidence was analyzed for neutrophil engraftment, platelet engraftment, GVHD, and TRM. Competing risks for engraftment were death, autologous recovery, and a second transplant. These and graft failure were competing risks for GVHD. TRM was defined as death not preceded by autologous recovery. EFS and OS were estimated using the Kaplan-Meier method. Death, autologous recovery, primary graft failure, or secondary graft failure were considered as events for EFS. Outcomes in the SC cohort were presented descriptively.
Results
Demographics and baseline characteristics
The characteristics of the 13 patients (4 male, 9 female) in the DC cohort are shown in Table 1. Median age was 13 (range, 3-17) years. All were African American with severe homozygous sickle cell disease (HbSS) and were eligible for transplant based on recurrent vaso-occlusive events (n = 8), acute chest syndrome (n = 3), or central nervous system risk factors (n = 6). Patient DC-20 had undergone reduced-intensity, haploidentical HSCT, whereas all others had undergone no previous transplant. Hydroxyurea was administered to 10 (77%) of the patients before enrollment. Ten patients (62%), including 8 in the DC and 2 in the SC cohorts received long-term transfusion therapy (simple transfusions, n = 8; erythrocytapheresis, n = 2) for amelioration of disease before transplantation. Other pretransplant comorbidities included avascular necrosis of the hip (n = 2), Wolff-Parkinson-White syndrome necessitating catheter ablation (n = 1), severe hepatobiliary disease (n = 1), and recurrent priapism (n = 1). The patients had a performance score of ≥80, and 7 (54%) were cytomegalovirus (CMV) seropositive at baseline.
The SC cohort included 1 female patient and 2 male patients, all African American, ages 12 to 17 years, 2 with HbSS, and one with HbS/β0 thalassemia (Table 1).
Graft characteristics
Patients in the DC cohort were infused with both omidubicel and an unmanipulated CBU. For CBUs used for omidubicel production, 92% (12 of 13) were 4-of-6 HLA matched with the recipient. For the unmanipulated CBUs, 69% (9 of 13) were 4-of-6 HLA matched with the recipient. No patients received a 6-of-6–matched unit. Allelic level matching (HLA A, B, C, and DRB1) between patients and the omidubicel unit was 7 of 8 (n = 1), 6 of 8 (n = 3), 5 of 8 (n = 3), 4 of 8 (n = 5), and 3 of 8 (n = 1); between patients and the unmanipulated unit, matching was 5 of 8 (n = 8), 4 of 8 (n = 4), and 2 of 8 (n = 1). The CBUs used for omidubicel production contained a median of 0.34 × 106 CD34+ cells per kilogram before expansion compared with 0.18 × 106 CD34+ cells per kilogram for the unmanipulated units. After ex vivo expansion, omidubicel contained a median of 12.3 × 106 CD34+ cells per kilogram (range, 2.55-19.51), reflecting a median 79-fold expansion (range, 36-110). The median CD3+ cell dose of the omidubicel unit, derived solely from the NF, was 4.19 × 106/kg (range, 1.85-29.37) and that of the unmanipulated unit was 5.43 × 106/kg (range, 3.52-9.01; data available for n = 12). The CD3+ cell dose in omidubicel was expected to be ∼30% lower after cryopreservation and thawing, based on previous data.16 Patients in the SC cohort underwent transplantation with omidubicel alone from 4 of 6 HLA-matched CBUs with TNC and CD34+ cell counts within the range of those used for DC patients (Figure 1; supplemental Table 1).
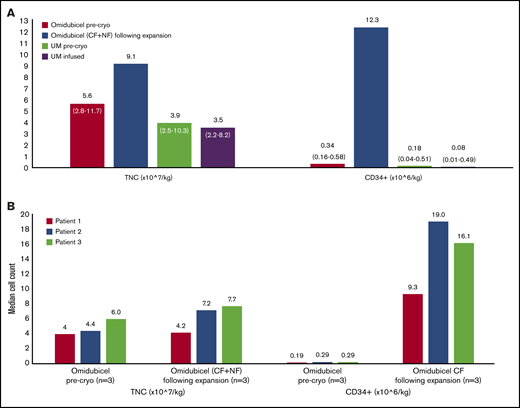
Graft characteristics. TNC dose and CD34+ cell dose for unmanipulated UCB units and before and after ex vivo expansion of the CBU for omidubicel. Precryopreservation values represent cell content as reported by the cord blood bank. (A) DC cohort (n = 13), median (range). (B) SC cohort (n = 3), individual patient results. Cryo, cryopreservation; UM, unmanipulated cord.
Graft characteristics. TNC dose and CD34+ cell dose for unmanipulated UCB units and before and after ex vivo expansion of the CBU for omidubicel. Precryopreservation values represent cell content as reported by the cord blood bank. (A) DC cohort (n = 13), median (range). (B) SC cohort (n = 3), individual patient results. Cryo, cryopreservation; UM, unmanipulated cord.
Clinical outcomes: DC cohort
Hematopoietic recovery.
All 13 patients initially attained engraftment of neutrophils at a median of 7 days (range, 6-20; 95% confidence interval [CI], 7-9). In 4 patients, ANC declined to <0.5 × 103/μL shortly after initial engraftment. Patient DC-3 attained engraftment on day 7 and had secondary graft failure on day 13 and died after a second allogeneic transplant. The other 3 patients with ANC decline had evidence of switching of chimerism from omidubicel to the unmanipulated unit, accompanied by neutrophil recovery at 15, 24, and 37 days after transplantation, without additional intervention. Overall, engraftment was sustained in 12 of 13 (92%) patients.
All patients had molecular evidence of predominantly omidubicel unit engraftment at day 7 (Figures 2,3-4). Of the 12 patients who attained engraftment, 10 (83%) switched donor chimerism from omidubicel to the unmanipulated unit at a median of 14 days. The cumulative incidence of donor cell chimerism was 100% (95% CI, 75-100). Overall, 2 patients had long-term engraftment with omidubicel, and 10 patients had it with the unmanipulated cord.

Chimerism: whole blood. Percentage of whole-blood chimerism from omidubicel (red) and unmanipulated UCB graft (blue) are shown in individual patients in the DC cohort (n = 13).
Chimerism: whole blood. Percentage of whole-blood chimerism from omidubicel (red) and unmanipulated UCB graft (blue) are shown in individual patients in the DC cohort (n = 13).

Chimerism: CD3+fraction. Percentage of CD3+ (T-cell) peripheral blood chimerism from omidubicel (red) and unmanipulated UCB graft (blue) are shown in individual patients in the DC cohort (n = 13).
Chimerism: CD3+fraction. Percentage of CD3+ (T-cell) peripheral blood chimerism from omidubicel (red) and unmanipulated UCB graft (blue) are shown in individual patients in the DC cohort (n = 13).
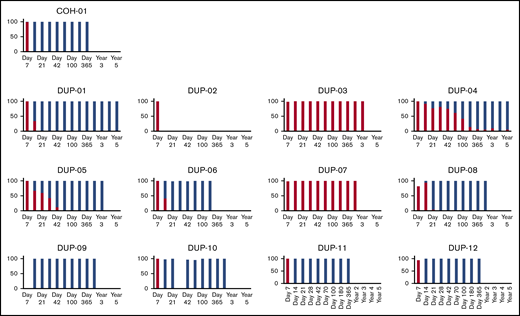
Chimerism: myeloid fraction. Percentage of myeloid peripheral blood chimerism from omidubicel (red) and unmanipulated UCB graft (blue) are shown in individual patients in the DC cohort (n = 13).
Chimerism: myeloid fraction. Percentage of myeloid peripheral blood chimerism from omidubicel (red) and unmanipulated UCB graft (blue) are shown in individual patients in the DC cohort (n = 13).
The degree of allelic level HLA mismatching of the UCB unit in patients was not predictive of long-term engraftment. Allelic matching between patients and the UCB unit with lasting engraftment was 6 of 8 (n = 1), 5 of 8 (n = 6), 4 of 8 (n = 4), and 2 of 8 (n = 1; supplemental Table 1). Of the 12 patients evaluable for engraftment beyond 100 days, 3 had received grafts of the better matched unit, 6 the less well-matched unit, and 3 the same level of match in both units.
Platelet engraftment was achieved at a median of 61 days (range, 33-375) with a cumulative incidence of platelet engraftment at day 100 of 61.5% (95% CI, 28-83).
Toxicity and adverse events.
Infusion of omidubicel was well tolerated. Three patients had a grade 3 event within 24 hours of infusion: 1 had a severe allergic reaction after the start of the unmanipulated CBU infusion (before omidubicel infusion), and 2 had pretransplant hypertension that improved after infusion.
Eleven patients (85%) had grade 2 and 3 posttransplant infections. The cumulative incidence was 31% at 6 months and 54% at 1 year, which was high (supplemental Table 2). Bacterial infections included Clostridium difficile enteritis (n = 5), UTI with Acinetobacter and Enterococcus (n = 1), Staphylococcus cellulitis (n = 1), biliary tract Stenotrophomonas (n = 1), and bacteremias (coagulase negative Staphylococcus, n = 3; Pseudomonas, n = 1; Enterococcus gallinarium, n = 1, vancomycin-resistant Enterococcus, n = 1; Enterobacter cloacae, n = 1; and Citrobacter and Acinetobacter, n = 1). CMV reactivation was reported in 4 patients and was cleared in all except patient DC-3, who had CMV positivity at death. Patient DC-11 received 2 courses of alemtuzumab for treatment of GVHD and developed CMV retinitis, which resolved after treatment with CMV virus-specific cytotoxic T lymphocytes and intravitreal therapy. Other viral infections included HHV6 viremia (n = 5), BK viruria with cystitis (n = 4), adenoviremia (n = 2), and herpes simplex virus (keratitis, n = 1; stomatitis, n = 1).
Two patients (15%) developed posterior reversible encephalopathy syndrome at days 43 and 95, which resolved with no sequelae. Other neurologic adverse events were acute subdural hemorrhage (n = 1) and busulfan-induced seizures (n = 1), both of which resolved without sequelae. The patient with subdural hemorrhage presented late, on day +340; did not have thrombocytopenia, hypertension, or magnetic resonance imaging/magnetic resonance angiographic evidence of cerebral vasculopathy or posterior reversible encephalopathy syndrome; and recovered with conservative management without sequelae. Thus, the etiology of this event is unknown.
GVHD.
Acute GVHD grades 2 to 4 were observed in 10 patients, 4 of whom had grades 3 and 4. The cumulative incidence of acute GVHD grades 2 to 4 at day 100 was 69.2% (95% CI, 34-88.2); grade 3 and 4 was 23.1% (95% CI, 5.1-48.6). The predominant organs involved were skin (n = 10), gut (n = 7), and liver (n = 1). Seven patients had chronic GVHD, and 4 had extensive chronic GVHD. The cumulative incidence of chronic GVHD at 6 and 12 months was 15% (95% CI, 2-40) and 46% (95% CI, 18-71), respectively. Patients with severe GVHD were treated with corticosteroids and other agents, including tacrolimus (n = 4), infliximab (n = 1), sirolimus (n = 1), mesenchymal stromal cells (n = 2), and alemtuzumab (n = 1). One patient died of liver GVHD. Resolution of GVHD symptoms allowed for discontinuation of immunosuppression in all 11 surviving patients at a median of 14 months (range, 7.9-22.9).
Immune reconstitution
Lymphoid immune recovery was monitored over time with 2-year data available for 9 patients (Figure 5). Median CD3, CD4, CD8, CD45RA+62L+ (naive T cells), CD4+CD25+ (T-Reg cells), CD19 (B cells), and CD16+56+ (natural killer cells) counts (cells per microliter) were 2580 (range, 1296-4094), 1338 (708-2194), 956 (336-1868), 1966 (1011-5258), and 215 (98-666), respectively, at 2 years after transplantation. All 11 surviving patients discontinued immunosuppression and immunoglobulin replacement, had normal IgG levels, and initiated vaccination.

Immune reconstitution. Cell counts (cells per microliter) for CD3, CD4, CD8, CD45RA+62L+, CD4+CD25+, CD19, and CD16+56+ cells at 100, 180, 365, and 730 days after transplantation in a subset of patients in the DC cohort (n = 9).
Immune reconstitution. Cell counts (cells per microliter) for CD3, CD4, CD8, CD45RA+62L+, CD4+CD25+, CD19, and CD16+56+ cells at 100, 180, 365, and 730 days after transplantation in a subset of patients in the DC cohort (n = 9).
Sickle cell disease–related parameters
The patients had a median HbS level of 20% before transplant, after planned transfusions. After HSCT, 10 of 11 surviving patients had no detection of HbS, and 1 patient had 40% HbS, consistent with sickle cell trait in the donor. The nontransfused hemoglobin and percentage of reticulocyte after transplantation were a median of 12.8 g/dL (range, 10.4-14.7) and 2.13% (range, 1.1-3.38), respectively. The median total bilirubin was 0.65 mg/dL (range, 0.4-1.4). All 11 surviving patients were reported to be free of SCD-related symptoms, with no reports of SCD-related events after transplant, including stroke, priapism, acute chest syndrome and vaso-occlusive pain crisis.
Survival
Eleven of 13 patients were alive and SCD free at follow-up. There was no TRM in the first 100 days and a 15% incidence of TRM during the first year. Twelve-month estimates of OS and EFS were both 84.6% (95% CI, 51.2-95.9). Two patients died after transplantation: patient DC-3 had secondary graft failure and died of infectious complications (disseminated adenovirus, CMV infections, and recurrent Enterococcus and Citrobacter bacteremia) 88 days after a second unrelated UCBT, which was 137 days after the first transplant. Patient DC-7 had a history of hepatobiliary disease before transplant and died of liver GVHD, multiple infections, and multisystem organ failure at 241 days after transplant.
SC cohort
All 3 patients in the SC cohort had donor-derived neutrophil engraftment (range, 7-9 days), with donor chimerism >90%. Toxicities included grade 3 acute GVHD in the gut resolving with treatment in patients SC-1 and SC-2, and veno-occlusive disease and renal failure in patient SC-1. Patient SC-3 developed secondary graft failure after initial engraftment, with aplasia, fevers, and CMV viremia. Immunosuppression was discontinued and autologous marrow infused (day +22), with full recovery of bone marrow function and resolution of CMV viremia. All 3 patients were alive at 1 year after transplant; patients SC-1 and SC-2 maintained full donor chimerism (100%), discontinued immunosuppression at 11 and 10 months, and had no measurable HbS through follow-up.
Discussion
In this study, we treated 16 pediatric patients with SCD with omidubicel in combination with an unmanipulated UCB unit (n = 13) and with a stand-alone omidubicel graft (n = 3). All surviving patients continued to demonstrate full donor chimerism with correction of HbS production, normalizing the hematological parameters without recurrence of SCD-related acute events. All 13 patients in the DC cohort had prompt engraftment with omidubicel, with long-term engraftment replaced by the unmanipulated CBU in 10 patients. All surviving patients (DC+SC) who attained engraftment achieved normal immune reconstitution by 1 to 2 years after transplant, including 4 patients with long-term omidubicel engraftment. Overall, 13 of 16 patients are alive with full donor engraftment without evidence of SCD after transplant.
Resistance to engraftment observed in patients with hemoglobinopathies undergoing HSCT has been attributed to compensatory bone marrow hyperactivity, the recipient’s chemotherapy-naive state, and alloimmunization by frequent blood transfusions. The cell dose of a standard UCB graft, 10- to 20-fold lower than other graft sources, is critical for engraftment, particularly in the hemoglobinopathy setting.9,22 Use of unrelated UCB donors is even more challenging because of lower cell doses and more HLA mismatching.23
Since 2005, several approaches to increase the cell dose in UCBT have been tested in the clinic. These include combining a UCB with another unit or third-party cells (eg, mesenchymal, haploidentical), or ex vivo expansion of UCB before infusion. However, prior approaches have had minimal impact on overall engraftment or survival.11-17,24,25 Horwitz et al demonstrated the safety and efficacy of ex vivo expansion of UCB with omidubicel in adults with HM, initially transplanting omidubicel with an unmanipulated cord unit, and more recently with an omidubicel stand-alone graft.18,20
As in the HM studies, we observed excellent expansion of CD34+ cells (∼80-fold median expansion) in the omidubicel CF. We believe this increase (median, 123.6 × 105/kg infused) facilitated very rapid and consistent early neutrophil engraftment with omidubicel cells (median, 7 days). However, unlike the adult HM phase I study,18 in which patients received a median CD34+ dose of 35 × 105/kg, the omidubicel graft was outcompeted by the unmanipulated UCB in the majority (10 of 12) of the patients with SCD. The reasons for this difference are not clear. Patients with SCD have more robust bone marrow cellularity compared with those with HM, as well as features of hyperinflammation (eg, vasculopathy), and are intrinsically more resistant to engraftment. One possible explanation is that there is a specific CD8+ T-cell–mediated “graft-versus-graft” immune reaction from the dominant graft against the nonengrafted unit.12,26 In addition, the omidubicel unit with an expected lower T-cell dose could have been disadvantaged immunologically. We did not study the CD8+ responses in the patients; however, there was a tendency toward higher CD3+ cell doses in the unmanipulated UCB.
Although the lack of persistence of the omidubicel graft in the setting of a competing unmanipulated UCB graft is intriguing, data for the use of omidubicel as a stand-alone graft indicate reassuringly consistent and durable engraftment in adults and adolescents with HM.20 Two patients in this study had engraftment of a stand-alone graft, although more data are needed to confirm consistent engraftment with a stand-alone graft in this population. The role of omidubicel in conferring long-term engraftment of UCB cannot be addressed in this study, but it has been shown to do so in studies of adults with HMs treated with omidubicel as a single graft source.
We retained the conventional myeloablative regimens for this study because of concerns of engraftment with reduced-intensity conditioning in the setting of UCBT in SCD.10 However, if future studies with omidubicel as a stand-alone graft in these patients continue to demonstrate high rates of durable engraftment, it may be possible to test omidubicel with reduced-intensity approaches in this population.
The high incidence of acute GVHD was a major limitation of this study and was probably due to the degree of mismatching necessary to find donors for African American patients and by the need to select UCB units with adequate TNC count and CD34 doses for omidubicel manufacturing. It is well known that African American patients have a lower probability of finding fully matched donors in the unrelated registries due to higher HLA diversity and lower volunteerism in that community. Thus, it was not surprising that the majority of the patients in this study (92%) had long-term engraftment with ≥3 allele-mismatched cords (≤5 of 8 or less), including 42% who attained engraftment with ≥4 allele-mismatched cords (≤4 of 8 or less). It is encouraging to note, however, that symptoms of GvHD resolved in all surviving patients who discontinued immunosuppression treatment. Our results demonstrate improved EFS with comparable incidence of severe acute or chronic GVHD compared with UCBT studies reported thus far for patients with sickle cell disease.9,10,27
Future approaches to decrease GVHD could include the use of closer HLA-matched units if the CD34 content required for omidubicel expansion could be reduced. Increasing cord blood inventories may also increase the likelihood of finding better matched units. It may be feasible to limit future studies to no more than 1 allele-mismatched donor (only 78 of 8 or 8 of 8 donors). Double UCBT is known to increase the risk of acute GvHD, and future protocols involving a single omidubicel graft may improve outcomes. Using single omidubicel graft may also increase the chance of using a better matched UCB donor.
Other potential interventions to reduce acute GVHD should be explored, such as the use of novel drugs (eg, abatacept) or in vivo T-cell depletion (eg, posttransplant cyclophosphamide [PT/Cy]).28,29 The preservation of early neutrophil engraftment with a lower incidence of GVHD overall could be explored using PT/Cy with an omidubicel graft. Alternatively, the omidubicel CF could be split to give a boost of myeloid cells after administration of PT/Cy. These interventions have the potential to influence engraftment and should be considered carefully.
The ex vivo expansion approach with UCB achieved complete donor chimerism and the elimination of sickle cell production in patients with the expected medical benefits. The potential barriers for future widespread adoption of this approach are the risks involved in myeloablative conditioning and the burden of GVHD. These risks should be assessed in the current context of alternative treatments, including haploidentical transplantation and autologous gene therapy. In the former, rejection risk has been traditionally high, although current trials with modified regimens appear promising.30-32 Autologous gene therapy has a clear advantage, given the short-term safety profile and lack of GVHD, but long-term efficacy remains in question and is being pursued in current studies.33 The uncertainty of long-term risks of genotoxicity is an important consideration during decision-making for curative therapies.34
In summary, this study demonstrated rapid and excellent engraftment with UCB in patients with SCD who have intrinsic resistance to engraftment, by means of ex vivo expansion with omidubicel technology. To date, enhanced engraftment has been demonstrated with omidubicel in 2 disorders with different risks of rejection: SCD (this study) and HM.18,20 This approach has potential applications in other disorders, such as bone marrow failure disorders.
Deidentified clinical details and protocol information may be obtained by e-mail request to the corresponding author (Suhag Parikh; suhag.parikh@emory.edu).
Acknowledgments
The authors thank all the members of the Duke Pediatric Blood and Marrow Transplantation Program, Gamida Cell, and The Emmes Corporation for their assistance with the study; patients and their families for their participation; Jennifer Baker for supervising the data collection; and Leonard Lionnet for providing medical writing support.
This study was supported by Gamida Cell.
Authorship
Contribution: J.K. conceptualized and designed the study, enrolled patients, critically reviewed and edited the manuscript, and provided comprehensive direction for the study; S.P. designed the study, enrolled patients, and wrote the first draft of the manuscript; E.G. wrote the protocol, monitored the conduct of the study, and edited the manuscript; A.S. acquired and analyzed data for the manuscript; and J.A.B. enrolled a patient in the study and edited the manuscript.
Conflict-of-interest disclosure: E.G. and A.S. are employees of Gamida Cell. The remaining authors declare no competing financial interests.
Correspondence: Suhag Parikh, Aflac Cancer and Blood Disorders Center, Children’s Healthcare of Atlanta, Emory University School of Medicine, 2015 Uppergate Dr, Atlanta, GA 30322; e-mail: suhag.parikh@emory.edu.

