

Key Points
Targeting codon-optimized FIX-Padua to platelets remarkably enhances platelet-FIX:C levels in primed FIXnull mice without anaphylaxis.
Platelet-targeted FIX-Padua gene therapy induces immune tolerance and provides sustained hyperfunctional platelet-FIX in primed FIXnull mice.

Abstract
Gene therapy may lead to a cure for hemophilia B (HB) if it is successful. Data from clinical trials using adeno-associated virus (AAV)–mediated liver-targeted FIX gene therapy are very encouraging. However, this protocol can be applied only to adults who do not have liver disease or anti-AAV antibodies, which occur in 30% to 50% of individuals. Thus, developing a protocol that can be applied to all HB patients is desired. Our previous studies have demonstrated that lentivirus-mediated platelet-specific FIX (2bF9) gene therapy can rescue bleeding diathesis and induce immune tolerance in FIXnull mice, but FIX expression was only ∼2% to 3% in whole blood. To improve the efficacy, we used a codon-optimized hyperfunctional FIX-Padua (2bCoF9R338L) to replace the 2bF9 cassette, resulting in 70% to 122% (35.08-60.77 mU/108 platelets) activity levels in 2bCoF9R338L-transduced FIXnull mice. Importantly, sustained hyperfunctional platelet-FIX expression was achieved in all 2bCoF9R338L-transduced highly immunized recipients with activity levels of 18.00 ± 9.11 and 9.36 ± 12.23 mU/108 platelets in the groups treated with 11 Gy and 6.6 Gy, respectively. The anti-FIX antibody titers declined with time, and immune tolerance was established after 2bCoF9R338L gene therapy. We found that incorporating the proteasome inhibitor bortezomib into preconditioning can help eliminate anti-FIX antibodies. The bleeding phenotype in 2bCoF9R338L-transduced recipients was completely rescued in a tail bleeding test and a needle-induced knee joint injury model once inhibitors dropped to undetectable. The hemostatic efficacy in 2bCoF9R338L-transduced recipients was further confirmed by ROTEM and thrombin generation assay (TGA). Together, our studies suggest that 2bCoF9R338L gene therapy can be a promising protocol for all HB patients, including patients with inhibitors.
Introduction
Hemophilia B (HB) is a genetic bleeding disorder resulting from a factor IX (FIX) deficiency.1 Protein replacement therapy is effective for the disease, but it is constrained by the short half-life of FIX, requiring frequent infusions.2-6 Furthermore, 5% of patients will develop neutralizing antibodies (inhibitors) against FIX,7,8 for which there is no effective approach for inducing immune tolerance.9 Moreover, anaphylactic reaction to the infused FIX protein in patients with inhibitors is a daunting problem that increases the risk of morbidity and mortality.7,10-14 Therefore, an effective protocol for treating patients with inhibitors is urgently needed.
Gene therapy is an alternative for HB treatment. Substantial progress in preclinical studies has been achieved in the last 2 decades.15-36 It has been shown that lentivirus (LV)- or adeno-associated virus (AAV)–mediated liver-targeted F9 gene transfer can reverse preexisting anti-FIX immunity and subsequently establish therapeutic levels of FIX in HB animal models,15,32 but 25% of inhibitor-prone mice were nonresponders with no FIX detectable after treatment.15 Clinical trials involving HB patients show that infusion of the AAV8 vector encoding codon-optimized FIX driven by a liver-specific promoter leads to sustained therapeutic levels of FIX expression.37-40 Furthermore, a combined effect of codon optimization and the gain-of-function FIX-Padua variant (R338L) can significantly enhance the efficacy of liver-targeted gene therapy in HB.41,42 These data are very encouraging, but an AAV-mediated liver-targeted protocol can be applied only to adults without liver disease or anti-AAV antibodies, which are present in 30% to 50% of the population.43-45 Thus, an alternative gene therapy approach is desired.
We have developed a platelet-specific gene therapy protocol for hemophiliacs that targets transgene expression to platelets under the control of the platelet-specific αIIb promoter.46-53 We have shown that platelet-specific FVIII expression (2bF8) can restore hemostasis in hemophilia A (HA) mice, even those with inhibitors.47,49,52 But platelet-specific FIX expression rescues bleeding diathesis only in HB mice without inhibitors54 because FIX does not have a protective carrier protein, unlike FVIII which is protected by von Willebrand factor (VWF).55-57 However, platelet-FIX gene therapy can induce FIX-specific immune tolerance in HB mice in the noninhibitor model.53 Here we explored platelet-targeted codon-optimized hyperfunctional FIX gene therapy for HB, even in mice with preexisting anti-FIX immunity.
Materials and methods
The following paragraphs briefly summarize the more detailed descriptions provided in the supplemental Data regarding antibodies and reagents, as well as methods and statistical analyses used in this study. FIX-deficient (FIXnull) mice in either a C57BL/6 background (Model 1) or in a B6-129S mixed background (Model 2) were used. The construct pWPT-2bF9 with wild-type human FIX (WT-hFIX) driven by the αIIb promoter was created as reported.54 The novel lentiviral vector, pWPT-2bCoF9R338L harboring a codon-optimized hFIX-Padua58,59 (CoF9R338L) directed by the αIIb promoter was constructed by replacing WT-hFIX in pWPT-2bF9 with CoF9R338L. 2bCoF9R338L and 2bF9 lentiviruses (LVs) were produced as previously reported.48,60,61
Sca-1+ cells isolated from FIXnull mice were transduced with 2bCoF9R338L or 2bF9 LV following the procedures previously described.53 Transduced cells were transplanted into each recipient preconditioned with either sublethal 6.6 Gy or lethal 11 Gy total body irradiation (TBI). For the inhibitor model, both donor and recipient FIXnull mice were preimmunized with recombinant hFIX (rhFIX) plus incomplete Freund’s adjuvant (IFA) to induce anti-FIX antibody development.
Quantitative analysis and assays
The 2bCoF9R338L transgene expression was determined by polymerase chain reaction (PCR) and quantitative real-time PCR (qRT-PCR) analysis. FIX expression levels were determined by a chromogenic assay for FIX activity (FIX:C) and by enzyme-linked immunosorbent assay (ELISA) for FIX antigen (FIX:Ag) on platelet lysates as previously described.54 To examine whether platelet-FIX is γ-carboxylated, we performed a barium sulfate (BaSO4) precipitation assay.62 Intracellular location of the neoprotein expression in 2bCoF9R338L-transduced platelets was determined by confocal microscopy, and the percentage of platelets that expressed hFIX in recipients was determined by fluorescence-activated cell sorting (FACS) as reported.53 The hemophilic bleeding phenotype in 2bCoF9R338L-transduced recipients was assessed by tail bleeding test, joint injury, ROTEM, and native whole-blood thrombin generation assay (nWB-TGA) as reported.46,54,63-66
FIX immune response studies
Anti-FIX inhibitor titers were quantified by Bethesda assay, and anti-FIX total immunoglobulin G (IgG) titers were determined by ELISA as reported.54 For the noninhibitor model, 8 months after hematopoietic stem cell (HSC) transplantation (HSCT), recipients were challenged with rhFIX plus IFA to determine whether immune tolerance was established after 2bCoF9R338L gene therapy. For the inhibitor model, when inhibitor titers dropped to undetectable levels, recipients were re-challenged with rhFIX by IV administration to determine whether immune tolerance was developed after 2bCoF9R338L gene therapy.
Results
Targeting codon-optimized FIX-Padua expression to platelets in FIXnull mice (the noninhibitor model)
Two strains of FIXnull mice, in either a C57BL/6 (Model 1) or a B6-129S (Model 2) background, were used to evaluate the efficacy of 2bCoF9R338L gene therapy (Figure 1A). 2bCoF9R338L provirus DNA was detected by PCR in all 2bCoF9R338L-transduced recipients (supplemental Figure 1). In Model 1, platelet-FIX:C and FIX:Ag levels in 2bCoF9R338L-transduced recipients were 61.29 ± 10.24 and 12.85 ± 5.68 mU/108 platelets at week 4, but they significantly decreased to 28.58 ± 14.5 and 4.38 ± 1.79 mU/108 platelets, respectively, at week 13 after HSCT (Figure 1B). The ratio of platelet-FIX:C to FIX:Ag was 5.16 to 6.38 (Figure 1C). In contrast, platelet-FIX expression in Model 2 was sustained in all transduced recipients throughout the study period, with platelet-FIX:C and FIX:Ag from 65.59 ± 10.66 and 9.77 ± 3.05 mU/108 platelets at week 5 to 66.38 ± 15.55 and 11.24 ± 2.69 mU/108 platelets, respectively, at week 57 after HSCT (Figure 1D). The ratio of platelet-FIX:C to FIX:Ag was 6.14 to 7.89 (Figure 1E).
![Targeting codon-optimized FIX-Padua expression to platelets in FIXnullmice (the noninhibitor model). Two strains of FIX-deficient (FIXnull) mice were used to test 2bCoF9R338L expression. FIX expression was introduced by 2bCoF9R338L LV transduction of Sca-1+ cells isolated from donors and transplanted into littermate recipients under a sublethal 6.6 Gy TBI. After HSCT and at least 4 weeks of BM reconstitution, blood samples were collected from recipients at various time points for assays. (A) Diagrams of experimental design. (Ai) Mice in Model 1 had a pure C57BL/6 background. (Aii) Mice in Model 2 had a C57BL/6-B6-129S mixed background. (B) Platelet-FIX expression in 2bCoF9R338L-transduced recipients in Model 1. Platelets were isolated and lysed in 0.5% CHAPS (the zwitterionic detergent, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate; MP Biomedicals). Functional platelet-FIX activity (Platelet-FIX:C) in platelet lysates was determined by a chromogenic assay. Platelet-FIX antigen (Platelet-FIX:Ag) levels in platelet lysates were quantified by enzyme-linked immunosorbent assay (ELISA). (C) The ratio of platelet-FIX:C to platelet-FIX:Ag in transduced recipients in Model 1. (D) Platelet-FIX expression in 2bCoF9R338L-transduced recipients in Model 2. (E) The ratio of platelet-FIX:C to platelet-FIX:Ag in 2bCoF9R338L-transduced recipients in Model 2. (F) Average platelet-FIX:C and platelet-FIX:Ag levels in 2bCoF9R338L-transduced recipients. Individual mice were analyzed at various time points during the study, and the average platelet-FIX level was calculated. Normal plasma from the International Society of Thrombosis and Hemostasis (ISTH) was used as a reference. (G) The average ratio of platelet-FIX:C to platelet-FIX:Ag in 2bCoF9R338L-transduced recipients. (H) Average copy number of 2bCoF9R338L proviral DNA in 2bCoF9R338L-transduced recipients. DNA was purified from leukocytes isolated from peripheral blood. The proviral DNA copy number was determined by quantitative real-time PCR (qRT-PCR). Individual mice were analyzed more than once during the study, and the average copy number was calculated. (I) Average platelet-FIX:C expression in sequential transplantation recipients from a representative experiment. BM mononuclear cells were isolated from the primary 2bCoF9R338L-tranduced recipient (1°), transplanted into secondary recipients (2°), and subsequently tertiary recipients (3°). Individual mice were analyzed more than once during the study, and the average level was calculated. Data are presented as mean ± standard deviation (SD). ****P < .0001. n.s., not statistically significant; Plt, platelet.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/5/10.1182_bloodadvances.2020004071/1/m_advancesadv2020004071f1.png?Expires=1767714949&Signature=dqD2XcYgNhLZ63YKGw2oGjihes-l3aB2NmHs638qK9dLS-OiNbWaAygGlkpRrUjdo5mDq-0-wIENVOgEzO15PUIl4XV9BpIRRtuGLJFd20ZuSmK9lDt1nrgANLp5pjgLB3HSo~cpKwMqX1CfZiEuvfrda8b3LEXP-0DE~-SHhXt21GaZzeSHRvbr2BbPxs~III8DeKDgB~Y3pl~jK2ePjIfxk1QqjI1SjAxOAM~B1gk3OVpPbGzGNP-YNpGuQ-vzfK4u4SJ8DSJIBYHcIDBi7FWFicXSesdVKgZalAxWInP9K3jDsnw4MWVcqnn0hkIxQtIZrap6RL3RWbhHFQ0OAA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Targeting codon-optimized FIX-Padua expression to platelets in FIXnullmice (the noninhibitor model). Two strains of FIX-deficient (FIXnull) mice were used to test 2bCoF9R338L expression. FIX expression was introduced by 2bCoF9R338L LV transduction of Sca-1+ cells isolated from donors and transplanted into littermate recipients under a sublethal 6.6 Gy TBI. After HSCT and at least 4 weeks of BM reconstitution, blood samples were collected from recipients at various time points for assays. (A) Diagrams of experimental design. (Ai) Mice in Model 1 had a pure C57BL/6 background. (Aii) Mice in Model 2 had a C57BL/6-B6-129S mixed background. (B) Platelet-FIX expression in 2bCoF9R338L-transduced recipients in Model 1. Platelets were isolated and lysed in 0.5% CHAPS (the zwitterionic detergent, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate; MP Biomedicals). Functional platelet-FIX activity (Platelet-FIX:C) in platelet lysates was determined by a chromogenic assay. Platelet-FIX antigen (Platelet-FIX:Ag) levels in platelet lysates were quantified by enzyme-linked immunosorbent assay (ELISA). (C) The ratio of platelet-FIX:C to platelet-FIX:Ag in transduced recipients in Model 1. (D) Platelet-FIX expression in 2bCoF9R338L-transduced recipients in Model 2. (E) The ratio of platelet-FIX:C to platelet-FIX:Ag in 2bCoF9R338L-transduced recipients in Model 2. (F) Average platelet-FIX:C and platelet-FIX:Ag levels in 2bCoF9R338L-transduced recipients. Individual mice were analyzed at various time points during the study, and the average platelet-FIX level was calculated. Normal plasma from the International Society of Thrombosis and Hemostasis (ISTH) was used as a reference. (G) The average ratio of platelet-FIX:C to platelet-FIX:Ag in 2bCoF9R338L-transduced recipients. (H) Average copy number of 2bCoF9R338L proviral DNA in 2bCoF9R338L-transduced recipients. DNA was purified from leukocytes isolated from peripheral blood. The proviral DNA copy number was determined by quantitative real-time PCR (qRT-PCR). Individual mice were analyzed more than once during the study, and the average copy number was calculated. (I) Average platelet-FIX:C expression in sequential transplantation recipients from a representative experiment. BM mononuclear cells were isolated from the primary 2bCoF9R338L-tranduced recipient (1°), transplanted into secondary recipients (2°), and subsequently tertiary recipients (3°). Individual mice were analyzed more than once during the study, and the average level was calculated. Data are presented as mean ± standard deviation (SD). ****P < .0001. n.s., not statistically significant; Plt, platelet.
Targeting codon-optimized FIX-Padua expression to platelets in FIXnullmice (the noninhibitor model). Two strains of FIX-deficient (FIXnull) mice were used to test 2bCoF9R338L expression. FIX expression was introduced by 2bCoF9R338L LV transduction of Sca-1+ cells isolated from donors and transplanted into littermate recipients under a sublethal 6.6 Gy TBI. After HSCT and at least 4 weeks of BM reconstitution, blood samples were collected from recipients at various time points for assays. (A) Diagrams of experimental design. (Ai) Mice in Model 1 had a pure C57BL/6 background. (Aii) Mice in Model 2 had a C57BL/6-B6-129S mixed background. (B) Platelet-FIX expression in 2bCoF9R338L-transduced recipients in Model 1. Platelets were isolated and lysed in 0.5% CHAPS (the zwitterionic detergent, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate; MP Biomedicals). Functional platelet-FIX activity (Platelet-FIX:C) in platelet lysates was determined by a chromogenic assay. Platelet-FIX antigen (Platelet-FIX:Ag) levels in platelet lysates were quantified by enzyme-linked immunosorbent assay (ELISA). (C) The ratio of platelet-FIX:C to platelet-FIX:Ag in transduced recipients in Model 1. (D) Platelet-FIX expression in 2bCoF9R338L-transduced recipients in Model 2. (E) The ratio of platelet-FIX:C to platelet-FIX:Ag in 2bCoF9R338L-transduced recipients in Model 2. (F) Average platelet-FIX:C and platelet-FIX:Ag levels in 2bCoF9R338L-transduced recipients. Individual mice were analyzed at various time points during the study, and the average platelet-FIX level was calculated. Normal plasma from the International Society of Thrombosis and Hemostasis (ISTH) was used as a reference. (G) The average ratio of platelet-FIX:C to platelet-FIX:Ag in 2bCoF9R338L-transduced recipients. (H) Average copy number of 2bCoF9R338L proviral DNA in 2bCoF9R338L-transduced recipients. DNA was purified from leukocytes isolated from peripheral blood. The proviral DNA copy number was determined by quantitative real-time PCR (qRT-PCR). Individual mice were analyzed more than once during the study, and the average copy number was calculated. (I) Average platelet-FIX:C expression in sequential transplantation recipients from a representative experiment. BM mononuclear cells were isolated from the primary 2bCoF9R338L-tranduced recipient (1°), transplanted into secondary recipients (2°), and subsequently tertiary recipients (3°). Individual mice were analyzed more than once during the study, and the average level was calculated. Data are presented as mean ± standard deviation (SD). ****P < .0001. n.s., not statistically significant; Plt, platelet.
The average platelet-FIX:C in Model 1 was significantly lower than that in Model 2 (Figure 1F), which was significantly higher than FIX expression in 2bF9-transduced recipients (supplemental Figure 2A). There were no significant differences in the average ratio of platelet-FIX:C to FIX:Ag or the average copy number of 2bCoF9R338L proviral DNA per cell between Model 1 and Model 2 (Figure 1G-H).
To further confirm whether long-term platelet-FIX expression was achieved after gene therapy, sequential transplantations were carried out from Model 2. Bone marrow (BM) cells collected from the primary (1°) recipients were transplanted into secondary (2°) and then tertiary (3°) recipients. The platelet-FIX:C expression levels in the sequential transplantation recipients were comparable to those in the primary recipients (Figure 1I).
Codon-optimized FIX-Padua expressed and stored in transduced platelets resulted in immune tolerance and phenotypic correction in FIXnull mice after gene therapy
To examine the transduction efficiency of 2bCoF9R338L gene therapy in FIXnull mice, we used FACS to analyze hFIX expression in platelets. The percentage of hFIX+ platelets in Model 2 after 2bCoF9R338L gene therapy was significantly higher than that in Model 1 (Figure 2A-B). The absolute levels of FIX:Ag and FIX:C in transduced platelets in the 2 models were comparable (Figure 2C) when calculated according to hFIX+ platelets measured by FACS. The percentages of hFIX+ platelets were similar between the 2bF9 and 2bCoF9R338L groups using Model 1 (supplemental Figure 2B).

Hyperfunctional FIX expressed in platelets induced immune tolerance in FIXnullmice that received 2bCoF9R338L-transduced HSCs (the noninhibitor model). (A) Flow cytometry analysis of platelets that were positive for hFIX protein expression in 2bCoF9R338L-transduced recipients. Platelets isolated from transduced recipients were permeabilized, stained with anti-hFIX–specific antibody (goat anti-hFIX primary antibody and donkey anti-goat IgG conjugated with Alexa-488 as a secondary antibody) and analyzed by flow cytometry. Platelets from FIXnull mice were used as a control in parallel. Shown are representative flow dot plots from recipients at 31 weeks after HSCT. (B) Summary data from flow cytometry analysis of hFIX+ platelets in 2bCoF9R338L-transduced FIXnull recipients. (C) Absolute levels of FIX expression in 2bCoF9R338L-transduced platelets. The absolute levels of FIX:C and FIX:Ag in 2bCoF9R338L-transduced platelets were calculated according to the platelet-FIX levels and the percentage of hFIX+ platelets in each recipient. (D) The incidence of anti-FIX inhibitor development in 2bCoF9R338L-transduced recipients after being challenged with rhFIX together with adjuvant. Eight months after transplantation, recipients were challenged with rhFIX at a dose of 200 U/kg in the presence of IFA twice with a 3-week interval. Two weeks after the second immunization, blood samples were collected, and plasma was isolated for a Bethesda assay to determine the inhibitor titers. FIXnull mice were immunized under the same protocol. (E) Anti-FIX inhibitor titers in 2bCoF9R338L-transduced recipients before and after challenge with rhFIX plus IFA. Data are presented as mean ± SD. **P < .01; ***P < .001. BU/mL, Bethesda units per mL; GPIIba, glycoprotein IIba; Post-Imm, post-immunization; Pre-Imm, pre-immunization.
Hyperfunctional FIX expressed in platelets induced immune tolerance in FIXnullmice that received 2bCoF9R338L-transduced HSCs (the noninhibitor model). (A) Flow cytometry analysis of platelets that were positive for hFIX protein expression in 2bCoF9R338L-transduced recipients. Platelets isolated from transduced recipients were permeabilized, stained with anti-hFIX–specific antibody (goat anti-hFIX primary antibody and donkey anti-goat IgG conjugated with Alexa-488 as a secondary antibody) and analyzed by flow cytometry. Platelets from FIXnull mice were used as a control in parallel. Shown are representative flow dot plots from recipients at 31 weeks after HSCT. (B) Summary data from flow cytometry analysis of hFIX+ platelets in 2bCoF9R338L-transduced FIXnull recipients. (C) Absolute levels of FIX expression in 2bCoF9R338L-transduced platelets. The absolute levels of FIX:C and FIX:Ag in 2bCoF9R338L-transduced platelets were calculated according to the platelet-FIX levels and the percentage of hFIX+ platelets in each recipient. (D) The incidence of anti-FIX inhibitor development in 2bCoF9R338L-transduced recipients after being challenged with rhFIX together with adjuvant. Eight months after transplantation, recipients were challenged with rhFIX at a dose of 200 U/kg in the presence of IFA twice with a 3-week interval. Two weeks after the second immunization, blood samples were collected, and plasma was isolated for a Bethesda assay to determine the inhibitor titers. FIXnull mice were immunized under the same protocol. (E) Anti-FIX inhibitor titers in 2bCoF9R338L-transduced recipients before and after challenge with rhFIX plus IFA. Data are presented as mean ± SD. **P < .01; ***P < .001. BU/mL, Bethesda units per mL; GPIIba, glycoprotein IIba; Post-Imm, post-immunization; Pre-Imm, pre-immunization.
To investigate the immune response in 2bCoF9R338L-transduced recipients, we monitored anti-FIX inhibitors after gene therapy, and no inhibitors were detected in transduced recipients. When recipients were challenged with rhFIX plus IFA, no Model 1 mice developed inhibitors; 3 of 7 Model 2 recipients developed low-titer inhibitors (1.1 to 2 Bethesda units per mL [BU/mL]), which subsequently disappeared within 4 weeks. The incidences of inhibitor development and the titers in transduced groups were significantly lower than in the control FIXnull group, in which all mice developed inhibitors with titers of 54.43 ± 47.54 BU/mL when the same immunization protocol was used (Figure 2D-E).
To assess the hemophilic phenotype in 2bCoF9R338L-transduced recipients, we used both in vivo models and ex vivo assays. In the 6-hour tail bleeding test, none of the FIXnull mice stopped bleeding within 6 hours, but all 2bCoF9R338L-transduced recipients did, with bleeding times of 2.5 ± 1.29 hours in Model 1 and 1.43 ± 0.79 hours in Model 2, which was not different compared with the WT control (Figure 3A). After the test, the percentages of remaining hemoglobin in 2bCoF9R338L-transduced recipients were significantly higher than those in FIXnull mice but not different compared with the WT control (Figure 3B).
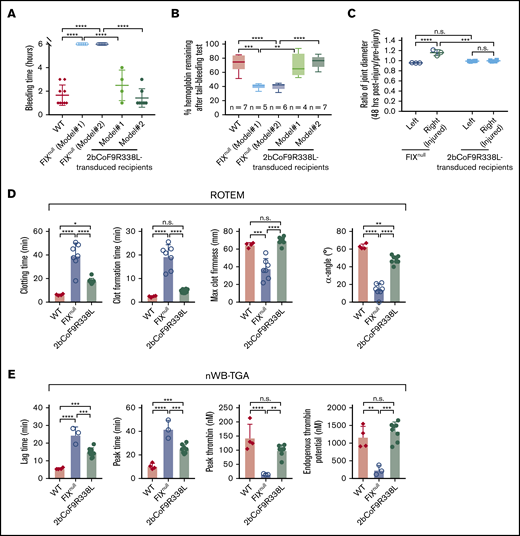
Assessment of the bleeding phenotype in FIXnullrecipients that received 2bCoF9R338L-transduced HSCs (the noninhibitor model). At least 6 months after transplantation, the bleeding phenotype was assessed by a 6-hour tail bleeding test and a needle-induced knee joint injury model. The functional hemostatic properties in whole blood were assessed by ROTEM and nWB-TGA analysis. For the tail bleeding test, the tail tip was clipped using a 1.6-mm diameter template. Animals were monitored hourly, and bleeding time was recorded. Fifty-microliter blood samples were collected for blood counts before and after the test. Hemoglobin levels before the test were defined as 100%. For the knee joint injury, a G30 × 1/2 needle was used to induce injury in the right knee, leaving the left knee uninjured as an intra-animal control. The diameter of the knee joint was measured by using a digital micro caliper before and 48 hours after injury. The diameter of the knee joint before the injury was defined as 1. For ROTEM and TGA analysis, blood samples were drawn from the vena cava. WT C57BL/6 and FIXnull mice served as controls. (A) Bleeding time from the tail bleeding test. (B) Percentage of hemoglobin remaining after the tail bleeding test. (C) The ratio of knee joint diameter obtained from 48 hours after compared with before the injury. (D) ROTEM analysis of whole blood. (E) TGA analysis of whole blood. Data are presented as mean ± SD except for panel B, which are presented in a box and whisker plot. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Assessment of the bleeding phenotype in FIXnullrecipients that received 2bCoF9R338L-transduced HSCs (the noninhibitor model). At least 6 months after transplantation, the bleeding phenotype was assessed by a 6-hour tail bleeding test and a needle-induced knee joint injury model. The functional hemostatic properties in whole blood were assessed by ROTEM and nWB-TGA analysis. For the tail bleeding test, the tail tip was clipped using a 1.6-mm diameter template. Animals were monitored hourly, and bleeding time was recorded. Fifty-microliter blood samples were collected for blood counts before and after the test. Hemoglobin levels before the test were defined as 100%. For the knee joint injury, a G30 × 1/2 needle was used to induce injury in the right knee, leaving the left knee uninjured as an intra-animal control. The diameter of the knee joint was measured by using a digital micro caliper before and 48 hours after injury. The diameter of the knee joint before the injury was defined as 1. For ROTEM and TGA analysis, blood samples were drawn from the vena cava. WT C57BL/6 and FIXnull mice served as controls. (A) Bleeding time from the tail bleeding test. (B) Percentage of hemoglobin remaining after the tail bleeding test. (C) The ratio of knee joint diameter obtained from 48 hours after compared with before the injury. (D) ROTEM analysis of whole blood. (E) TGA analysis of whole blood. Data are presented as mean ± SD except for panel B, which are presented in a box and whisker plot. *P < .05; **P < .01; ***P < .001; ****P < .0001.
When animals were challenged with a needle-induced joint injury, the diameter of injured joints (right knee) in FIXnull mice increased 1.159-fold ± 0.059-fold 48 hours after injury, which was significantly larger compared with the uninjured left knee control (0.957 ± 0.005-fold) (Figure 3C). Notably, in 2bCoF9R338L-transduced recipients, there was no significant change in the diameter of the injured joint after injury, and the ratio of diameter after the injury to the diameter at baseline was comparable to that in the uninjured intra-animal control (Figure 3C), demonstrating that platelet-CoF9R338L expression can effectively prevent joint bleeding.
The hemostatic efficacy of 2bCoF9R338L gene therapy was further assessed by ROTEM and nWB-TGA analysis. As shown in Figure 3D, all parameters in ROTEM, including whole-blood clotting time (CT), clot formation time (CFT), maximum clot firmness (MCF), and α-angle, in the 2bCoF9R338L group were significantly improved compared with the FIXnull group. There were no differences in CFT and MCF between the 2bCoF9R338L and WT groups, but CT was longer and α-angle was lower in the 2bCoF9R338L group compared with the WT group. When nWB-TGA was conducted, all parameters, including lag time (LT), peak time (PT), peak thrombin (PTh), and endogenous thrombin potential (ETP), in the 2bCoF9R338L group were also significantly improved compared with the FIXnull group (Figure 3E). There was no difference in PTh and ETP between the 2bCoF9R338L and WT groups, but LT and PT were longer in the 2bCoF9R338L group compared with the WT group (Figure 3E). Taken together, these results demonstrate that targeting codon-optimized FIX-Padua expression by lentiviral gene delivery to HSCs can introduce sustained hyperfunctional FIX expression in platelets, resulting in phenotypic correction and immune tolerance induction in HB mice.
Targeting codon-optimized FIX-Padua expression to platelets in rhFIX-primed FIXnull mice (the inhibitor model)
To investigate the feasibility of 2bCoF9R338L gene therapy for the inhibitor model, we used B6-129S FIXnull mice. 2bCoF9R338L-transduced cells were transplanted into rhFIX-primed recipients preconditioned with TBI at either 11 Gy or 6.6 Gy (Figure 4A). After 4 weeks of BM reconstitution, we started to analyze the animals. 2bCoF9R338L proviral DNA was detected by PCR in all transduced recipients (supplemental Figure 1), demonstrating the viability of 2bCoF9R338L genetically modified cells in primed FIXnull mice under either myeloablative or nonmyeloablative preconditioning. The average copy number per cell of 2bCoF9R338L in transduced recipients was similar in the 11-Gy and 6.6-Gy groups (Figure 4B-C). The whole-blood counts in transduced recipients were comparable to those in untransduced transplanted (UnTrans) controls (supplemental Figure 3A-C), demonstrating that platelet-specific FIX-Padua expression does not have an impact on blood cell reconstitution.
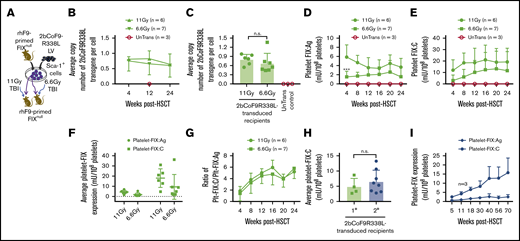
Targeting codon-optimized FIX-Padua expression to highly primed FIXnullmice (the inhibitor model). Both donor and recipient FIXnull mice were immunized with rhFIX at 200 U/kg in the presence of IFA twice with a 3-week interval to induce the development of anti-FIX antibodies. Two weeks after the second immunization, anti-FIX inhibitor titers were determined by Bethesda assay, and anti-FIX total IgG titers were determined by ELISA. Sca-1+ cells isolated from FIX-primed donors were transduced with 2bCoF9R338L LV and transplanted into FIX-primed recipients under either a lethal 11-Gy or a sublethal 6.6-Gy TBI. Four weeks after HSCT, blood samples were collected from various time points for assays. (A) Diagram of experimental design. FIXnull mice with a B6-129S mixed background were used. (B) Average copy number of 2bCoF9R338L proviral DNA per cell in transduced FIX-primed recipients during the study period. The copy number was determined by qRT-PCR. (C) Average copy number of 2bCoF9R338L proviral DNA per cell in each rhFIX-primed recipient. Individual mice were analyzed more than once during the study, and the average copy number was calculated. (D) Platelet-FIX:Ag expression levels in 2bCoF9R338L-transduced FIX-primed recipients during the study period. Platelets were isolated and lysed in 0.5% CHAPS. Platelet-FIX:Ag levels in platelet lysates were determined by ELISA. (E) Platelet-FIX activity (platelet-FIX:C) levels in 2bCoF9R338L-transduced FIX-primed recipients during the study period. Platelet-FIX:C levels in platelet lysates were quantified by a chromogenic assay. (F) Average platelet-FIX expression in 2bCoF9R338L-transduced FIX-primed recipients. Individual mice were analyzed at various time points, and the average FIX expression level was calculated. (G) The ratio of platelet-FIX:C to platelet-FIX:Ag during the study period. (H) Average platelet-FIX:C expression levels in sequential transplantation recipients. Four primary (1°) recipients from the 6.6-Gy group that received 2bCoF9R338L-transduced Sca-1+ cells were euthanized at 63 weeks after HSCT, and BM mononuclear cells isolated from the 1° recipients were transplanted into the 2° recipients that were also primed with rhFIX plus IFA and preconditioned with 6.6 Gy TBI plus bortezomib 1 mg/kg by IV administration. Platelet-FIX:C levels in recipients were monitored. Individual mice were analyzed more than once, and the average FIX expression level was calculated. (I) Sustained FIX expression in the 2° recipients during the study period. Data are presented as mean ± SD. ***P < .001.
Targeting codon-optimized FIX-Padua expression to highly primed FIXnullmice (the inhibitor model). Both donor and recipient FIXnull mice were immunized with rhFIX at 200 U/kg in the presence of IFA twice with a 3-week interval to induce the development of anti-FIX antibodies. Two weeks after the second immunization, anti-FIX inhibitor titers were determined by Bethesda assay, and anti-FIX total IgG titers were determined by ELISA. Sca-1+ cells isolated from FIX-primed donors were transduced with 2bCoF9R338L LV and transplanted into FIX-primed recipients under either a lethal 11-Gy or a sublethal 6.6-Gy TBI. Four weeks after HSCT, blood samples were collected from various time points for assays. (A) Diagram of experimental design. FIXnull mice with a B6-129S mixed background were used. (B) Average copy number of 2bCoF9R338L proviral DNA per cell in transduced FIX-primed recipients during the study period. The copy number was determined by qRT-PCR. (C) Average copy number of 2bCoF9R338L proviral DNA per cell in each rhFIX-primed recipient. Individual mice were analyzed more than once during the study, and the average copy number was calculated. (D) Platelet-FIX:Ag expression levels in 2bCoF9R338L-transduced FIX-primed recipients during the study period. Platelets were isolated and lysed in 0.5% CHAPS. Platelet-FIX:Ag levels in platelet lysates were determined by ELISA. (E) Platelet-FIX activity (platelet-FIX:C) levels in 2bCoF9R338L-transduced FIX-primed recipients during the study period. Platelet-FIX:C levels in platelet lysates were quantified by a chromogenic assay. (F) Average platelet-FIX expression in 2bCoF9R338L-transduced FIX-primed recipients. Individual mice were analyzed at various time points, and the average FIX expression level was calculated. (G) The ratio of platelet-FIX:C to platelet-FIX:Ag during the study period. (H) Average platelet-FIX:C expression levels in sequential transplantation recipients. Four primary (1°) recipients from the 6.6-Gy group that received 2bCoF9R338L-transduced Sca-1+ cells were euthanized at 63 weeks after HSCT, and BM mononuclear cells isolated from the 1° recipients were transplanted into the 2° recipients that were also primed with rhFIX plus IFA and preconditioned with 6.6 Gy TBI plus bortezomib 1 mg/kg by IV administration. Platelet-FIX:C levels in recipients were monitored. Individual mice were analyzed more than once, and the average FIX expression level was calculated. (I) Sustained FIX expression in the 2° recipients during the study period. Data are presented as mean ± SD. ***P < .001.
The level of platelet-FIX:Ag expression in the 11-Gy group at week 4 was significantly higher than in the 6.6-Gy group, but it was not different at other time points (Figure 4D). Platelet-FIX:C expression levels seem to be lower in both the 11-Gy and 6.6-Gy groups at 4 weeks after gene therapy, but there were no significant differences among time points within each group or between groups (Figure 4E). The average platelet-FIX:Ag and platelet-FIX:C levels were similar in the 11-Gy and 6.6-Gy groups (Figure 4F). The ratios of platelet-FIX:C to FIX:Ag in both groups were comparable, starting at around 2 at week 4 and increasing to 6 at week 16 (Figure 4G).
To confirm whether long-term platelet-FIX expression can be achieved in the setting of preexisting immunity, even under nonmyeloablative preconditioning, sequential transplantation was carried out by transplanting BM cells from primary recipients from the 6.6-Gy group into secondary rhFIX-primed FIXnull mice preconditioned with 6.6 Gy plus bortezomib. Platelet-FIX:C expression levels in the 2° recipients were similar to those obtained from the 1° recipients (Figure 4H) and sustained (Figure 4I) for a total of 2.55 years during our study period (63 weeks for 1° and 70 weeks for 2° recipients).
For comparison, we used 2bF9LV that harbors WT-hFIX to introduce platelet-FIX expression in rhFIX-primed FIXnull mice with either 11 Gy or 6.6 Gy TBI. All recipients had detectable levels of platelet-FIX:C expression at week 4 after 2bF9 gene therapy with levels of 2.31 ± 3.69 and 0.77 ± 0.31 mU/108 platelets, respectively, but 40% in the 11-Gy group and 75% in the 6.6-Gy group dropped to undetectable levels by 18 weeks after HSCT (supplemental Figure 4A-C). Together, these data demonstrate that LV-mediated platelet-specific gene delivery of 2bCoF9R338L can effectively introduce sustained hyperfunctional platelet-FIX expression even in highly primed FIXnull mice.
Hyperfunctional FIX-Padua expressed and stored in transduced platelets is γ-carboxylated
To examine the intracellular location of hFIX-Padua expression in platelets, we used confocal microscopy. hFIX protein was detected in 2bCoF9R338L-transduced platelets (Figure 5A,D) and colocalized with endogenous VWF protein in platelet α-granules (Figure 5C,F; shown in yellow). Platelets of UnTrans were positive for VWF but negative for hFIX protein as expected (Figure 5G-I). FACS analysis showed that 21.93% ± 11.6% of platelets were positive for hFIX in the 11-Gy group, which was not significantly different compared with the 6.6-Gy group (Figure 5J-K). There were no differences in the absolute hFIX:Ag and hFIX:C expression levels in transduced platelets between the 11-Gy and 6.6-Gy groups when calculated on the basis of the percentage of hFIX+carboxylated, we used BaSO4 to treat platelet lysates from 2bCoF9R338L- and 2bF9-transduced recipients. As shown in Figure 5M, no FIX:Ag remained in the supernatants of BaSO4-treated platelet lysates from either 2bF9- or 2bCoF9R338L-transduced recipients, in which various levels of platelet-FIX:Ag (0.69-15.62 mU/108 platelets) were added. As expected, nearly 100% of FIX:Ag was precipitated in rhFIX control supernatants after BaSO4 precipitation (Figure 5N). These data demonstrate that FIX expressed in platelets is γ-carboxylated.

Hyperfunctional FIX-Padua expressed and stored in platelets was fully γ-carboxylated. Intracellular location of codon-optimized FIX-Padua in 2bCoF9R338L-transduced platelets of FIX-primed FIXnull mice was examined by immunofluorescent confocal microscopy. The percentage of platelets that were positive for hFIX protein was determined by flow cytometry. To evaluate whether ectopic expression of codon-optimized FIX-Padua in platelets is fully γ-carboxylated, platelet lysates from 2bCoF9R338L-transduced recipients at various time points with varying FIX:Ag levels (0.69 to 15.62 mU/108 platelets) were treated with or without 10% BaSO4. The remaining FIX in supernatants after BaSO4 precipitation was determined by ELISA. Various amounts of rhFIX were treated with BaSO4 as controls in parallel. (A-I) Immunofluorescent confocal microscopy. hFIX was stained with a primary goat anti-hFIX antibody and a secondary donkey anti-goat IgG conjugated with Alexa-488 (in green). Endogenous VWF protein was stained with a rabbit anti-human VWF antibody that crosses with mouse VWF and a secondary donkey anti-rabbit IgG conjugated with Alexa-594 (in red). Scale bars, 10 μm. (J) Representative dot plots from flow cytometry analysis of hFIX+ platelets. hFIX was stained with a primary goat anti-hFIX antibody and a secondary donkey anti-goat IgG conjugated with Alexa-488. Platelets were identified using anti-GPIIb antibody directly conjugated with phycoerythrin. (K) Summary data from flow cytometry analysis of hFIX+ platelets in 2bCoF9R338L-transduced rhFIX-primed FIXnull recipients at weeks 20 to 28 after transplantation. (L) Absolute levels of FIX expression in 2bCoF9R338L-transduced platelets in rhFIX-primed recipients. The absolute levels of FIX:C and FIX:Ag in 2bCoF9R338L-transduced platelets were calculated according to the platelet-FIX levels and the percentage of hFIX+ platelets in each recipient. (M) Platelet-FIX:Ag in the supernatants of platelet lysates from 2bCoF9R338L- or 2bF9-transduced recipients after BaSO4 precipitation. Platelet-FIX:Ag levels in platelet lysates without BaSO4 treatment were defined as 100%. (N) FIX:Ag in the supernatants of rhFIX after BaSO4 precipitation. FIX:Ag levels in supernatants of rhFIX without BaSO4 treatment were defined as 100%. Data are presented as mean ± SD.
Hyperfunctional FIX-Padua expressed and stored in platelets was fully γ-carboxylated. Intracellular location of codon-optimized FIX-Padua in 2bCoF9R338L-transduced platelets of FIX-primed FIXnull mice was examined by immunofluorescent confocal microscopy. The percentage of platelets that were positive for hFIX protein was determined by flow cytometry. To evaluate whether ectopic expression of codon-optimized FIX-Padua in platelets is fully γ-carboxylated, platelet lysates from 2bCoF9R338L-transduced recipients at various time points with varying FIX:Ag levels (0.69 to 15.62 mU/108 platelets) were treated with or without 10% BaSO4. The remaining FIX in supernatants after BaSO4 precipitation was determined by ELISA. Various amounts of rhFIX were treated with BaSO4 as controls in parallel. (A-I) Immunofluorescent confocal microscopy. hFIX was stained with a primary goat anti-hFIX antibody and a secondary donkey anti-goat IgG conjugated with Alexa-488 (in green). Endogenous VWF protein was stained with a rabbit anti-human VWF antibody that crosses with mouse VWF and a secondary donkey anti-rabbit IgG conjugated with Alexa-594 (in red). Scale bars, 10 μm. (J) Representative dot plots from flow cytometry analysis of hFIX+ platelets. hFIX was stained with a primary goat anti-hFIX antibody and a secondary donkey anti-goat IgG conjugated with Alexa-488. Platelets were identified using anti-GPIIb antibody directly conjugated with phycoerythrin. (K) Summary data from flow cytometry analysis of hFIX+ platelets in 2bCoF9R338L-transduced rhFIX-primed FIXnull recipients at weeks 20 to 28 after transplantation. (L) Absolute levels of FIX expression in 2bCoF9R338L-transduced platelets in rhFIX-primed recipients. The absolute levels of FIX:C and FIX:Ag in 2bCoF9R338L-transduced platelets were calculated according to the platelet-FIX levels and the percentage of hFIX+ platelets in each recipient. (M) Platelet-FIX:Ag in the supernatants of platelet lysates from 2bCoF9R338L- or 2bF9-transduced recipients after BaSO4 precipitation. Platelet-FIX:Ag levels in platelet lysates without BaSO4 treatment were defined as 100%. (N) FIX:Ag in the supernatants of rhFIX after BaSO4 precipitation. FIX:Ag levels in supernatants of rhFIX without BaSO4 treatment were defined as 100%. Data are presented as mean ± SD.
Platelet-specific codon-optimized FIX-Padua gene therapy can induce immune tolerance in FIXnull mice with preexisting immunity
To investigate the immune responses in rhFIX-primed FIXnull mice after 2bCoF9R338L gene therapy, we monitored both anti-FIX inhibitors and total IgG titers in recipients. Inhibitor and total IgG titers declined with time after HSCT in both transduced and untransduced recipients (Figure 6A-B). The half-life of inhibitor disappearance time in the 11-Gy group was significantly shorter than that in the 6.6-Gy and UnTrans control groups (Figure 6C), but there were no differences in anti-FIX total IgG titers among groups (Figure 6D). Importantly, no anaphylaxis occurred in any FIX-primed HB mice after platelet gene therapy.
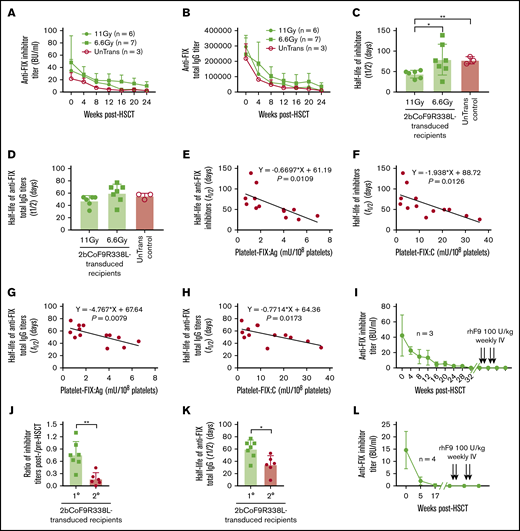
Immune responses in 2bCoF9R338L-transduced rhFIX-primed recipients. Anti-FIX antibody titers in rhFIX-primed FIXnull mice were monitored monthly after receiving 2bCoF9R338L-transduced HSCs. rhFIX-primed FIXnull mice preconditioned with 6.6 Gy TBI and transplanted with untransduced HSCs were used as controls in parallel. Anti-FIX inhibitor titers were determined by Bethesda assay, and anti-FIX total IgG titers were determined by ELISA. (A) Anti-hFIX inhibitor titers in 2bCoF9R338L-transduced rhFIX-primed FIXnull recipients. (B) Anti-hFIX total IgG titers in 2bCoF9R338L-transduced rhFIX-primed FIXnull recipients. (C) The half-life of anti-FIX inhibitor disappearance time. (D) The half-life of anti-FIX total IgG disappearance time. (E) The correlation between platelet-FIX:Ag levels and half-lives of anti-FIX inhibitor disappearance time in 2bCoF9R338L-transduced rhFIX-primed recipients. (F) The correlation between platelet-FIX:C levels and half-lives of anti-FIX inhibitor disappearance time in 2bCoF9R338L-transduced rhFIX-primed recipients. (G) The correlation between platelet-FIX:Ag levels and half-lives of anti-FIX total IgG disappearance time in 2bCoF9R338L-transduced rhFIX-primed recipients. (H) The correlation between platelet-FIX:C levels and half-lives of anti-FIX total IgG disappearance time in 2bCoF9R338L-transduced rhFIX-primed recipients. (I) Immune tolerance was established in 2bCoF9R338L-transduced rhFIX-primed primary recipients. When anti-FIX inhibitor titers dropped to undetectable, animals were challenged with rhFIX at 100 U/kg per week by IV administration for a total of 4 times. One week after the second and fourth re-challenge, blood samples were collected for the inhibitor assay. (J) Comparison of the changes in inhibitor titers in primary vs secondary recipients 1 month after to before HSCT. (K) Comparison of the half-life of anti-FIX total IgG disappearance time in primary vs secondary 2bCoF9R338L-transduced recipients. (L) Immune tolerance developed in 2bCoF9R338L-transduced secondary primed FIXnull recipients. Data are presented as mean ± SD. *P < .05; **P < .01.
Immune responses in 2bCoF9R338L-transduced rhFIX-primed recipients. Anti-FIX antibody titers in rhFIX-primed FIXnull mice were monitored monthly after receiving 2bCoF9R338L-transduced HSCs. rhFIX-primed FIXnull mice preconditioned with 6.6 Gy TBI and transplanted with untransduced HSCs were used as controls in parallel. Anti-FIX inhibitor titers were determined by Bethesda assay, and anti-FIX total IgG titers were determined by ELISA. (A) Anti-hFIX inhibitor titers in 2bCoF9R338L-transduced rhFIX-primed FIXnull recipients. (B) Anti-hFIX total IgG titers in 2bCoF9R338L-transduced rhFIX-primed FIXnull recipients. (C) The half-life of anti-FIX inhibitor disappearance time. (D) The half-life of anti-FIX total IgG disappearance time. (E) The correlation between platelet-FIX:Ag levels and half-lives of anti-FIX inhibitor disappearance time in 2bCoF9R338L-transduced rhFIX-primed recipients. (F) The correlation between platelet-FIX:C levels and half-lives of anti-FIX inhibitor disappearance time in 2bCoF9R338L-transduced rhFIX-primed recipients. (G) The correlation between platelet-FIX:Ag levels and half-lives of anti-FIX total IgG disappearance time in 2bCoF9R338L-transduced rhFIX-primed recipients. (H) The correlation between platelet-FIX:C levels and half-lives of anti-FIX total IgG disappearance time in 2bCoF9R338L-transduced rhFIX-primed recipients. (I) Immune tolerance was established in 2bCoF9R338L-transduced rhFIX-primed primary recipients. When anti-FIX inhibitor titers dropped to undetectable, animals were challenged with rhFIX at 100 U/kg per week by IV administration for a total of 4 times. One week after the second and fourth re-challenge, blood samples were collected for the inhibitor assay. (J) Comparison of the changes in inhibitor titers in primary vs secondary recipients 1 month after to before HSCT. (K) Comparison of the half-life of anti-FIX total IgG disappearance time in primary vs secondary 2bCoF9R338L-transduced recipients. (L) Immune tolerance developed in 2bCoF9R338L-transduced secondary primed FIXnull recipients. Data are presented as mean ± SD. *P < .05; **P < .01.
In 2bCoF9R338L-transduced recipients, the half-lives of anti-FIX inhibitor and total IgG disappearance were negatively correlated with platelet-FIX:Ag and platelet-FIX:C levels (Figure 6E-H). Furthermore, an equal amount of FIX:C in platelet lysates from either 2bF9- or 2bCoF9R338L-transduced recipients could neutralize the same amount of inhibitors, but the same amount of FIX:Ag from 2bCoF9R338L-transduced platelets showed a greater capability to neutralize the anti-FIX inhibitors (supplemental Figure 5). To investigate whether immune tolerance was induced in 2bCoF9R338L-transduced recipients after their inhibitor titers dropped to undetectable levels, animals were re-challenged with rhFIX at 100 U/kg per week by IV administration for 4 weeks. Remarkably, the inhibitor titers remained undetectable after rhFIX rechallenge (Figure 6I). In contrast, all FIXnull mice (n = 4) that were immunized with 2 doses of rhFIX plus IFA and confirmed to have developed inhibitors died within 4 hours when re-challenged with a single dose of rhFIX at 100 U/kg by IV administration (data not shown).
It has been shown that the proteasome inhibitor bortezomib can deplete plasma cells, reducing autoantibody production in autoimmune disease models.67-70 Here, we evaluated whether bortezomib combined with TBI would facilitate the elimination of anti-FIX antibodies in the inhibitor model. We added bortezomib to 6.6-Gy TBI as preconditioning for 2bCoF9R338L gene therapy in the secondary recipients. We found that inhibitor titers decreased faster in the secondary recipients preconditioned with 6.6 Gy plus bortezomib compared with the primary recipients under 6.6 Gy TBI (Figure 6J). The half-life of anti-FIX total IgG in the secondary recipients was significantly shorter than that in the primary recipients (Figure 6K). As with the primary recipients, once inhibitor titers dropped to undetectable levels in the secondary transplantation recipients, no inhibitors were detected, even after 4 doses of rhFIX rechallenge, demonstrating that immune tolerance was also established in the secondary transplantation recipients. Together, these data demonstrate that platelet-specific CoF9R338L expression does not trigger an anti-FIX memory immune response. Instead, 2bCoF9R338L gene therapy can circumvent anaphylaxis and induce immune tolerance in primed FIXnull mice.
Phenotypic correction assessment in primed FIXnull mice after 2bCoF9R338L gene therapy
Because the clinical efficacy of platelet-FIX is limited in FIXnull mice with inhibitors,54 we did not assess the bleeding phenotype in recipients until inhibitor titers dropped to undetectable levels. When a 6-hour tail bleeding test was used to evaluate the clinical efficacy of 2bCoF9R338L gene therapy in primed mice, the bleeding times in the 11-Gy and 6.6-Gy groups were similar to those in the WT control (Figure 7A). The remaining hemoglobin levels after the test in 2bCoF9R338L-transduced groups were comparable to the WT group but were significantly higher than in the FIXnull control (Figure 7B).
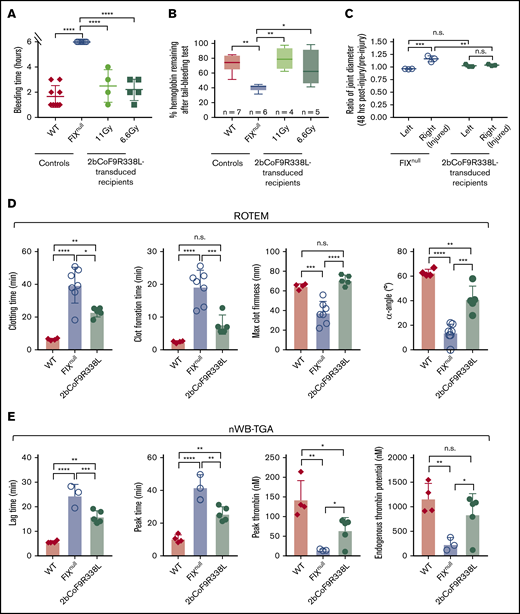
Assessment of the bleeding phenotype in 2bCoF9R338L-transduced rhFIX-primed recipients. The tail bleeding test and the needle-induced joint injury were performed on recipients at least 6 months after HSCT once their inhibitor titers dropped to undetectable. ROTEM and nWB-TGA were performed on blood drawn from the vena cava (terminal experiment). For the tail bleeding test, the tail tip was transected using a 1.6-mm diameter template. Animals were monitored hourly, and bleeding time was recorded. Fifty-microliter blood samples were collected for blood counts before and after the test. Hemoglobin levels before the test were defined as 100%. For the knee joint injury, a G30 × 1/2 needle was used to induce injury in the right knee, leaving the left knee uninjured as an intra-animal control. The diameter of the knee joint was measured using a digital micro caliper before and 48 hours after injury. The diameter of the knee joint before the injury was defined as 1. (A) Bleeding time from the tail bleeding test. (B) The percentage of hemoglobin remaining after the tail bleeding test. (C) The needle-induced knee joint injury. (D) ROTEM analysis of whole blood. (E) TGA analysis of whole blood. WT and FIXnull mice served as controls. Data are presented as mean ± SD except for panel B, which are presented in a box and whisker plot. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Assessment of the bleeding phenotype in 2bCoF9R338L-transduced rhFIX-primed recipients. The tail bleeding test and the needle-induced joint injury were performed on recipients at least 6 months after HSCT once their inhibitor titers dropped to undetectable. ROTEM and nWB-TGA were performed on blood drawn from the vena cava (terminal experiment). For the tail bleeding test, the tail tip was transected using a 1.6-mm diameter template. Animals were monitored hourly, and bleeding time was recorded. Fifty-microliter blood samples were collected for blood counts before and after the test. Hemoglobin levels before the test were defined as 100%. For the knee joint injury, a G30 × 1/2 needle was used to induce injury in the right knee, leaving the left knee uninjured as an intra-animal control. The diameter of the knee joint was measured using a digital micro caliper before and 48 hours after injury. The diameter of the knee joint before the injury was defined as 1. (A) Bleeding time from the tail bleeding test. (B) The percentage of hemoglobin remaining after the tail bleeding test. (C) The needle-induced knee joint injury. (D) ROTEM analysis of whole blood. (E) TGA analysis of whole blood. WT and FIXnull mice served as controls. Data are presented as mean ± SD except for panel B, which are presented in a box and whisker plot. *P < .05; **P < .01; ***P < .001; ****P < .0001.
When the needle-induced joint injury model was used to assess the efficacy of 2bCoF9R338L gene therapy in preventing joint bleeds in primed FIXnull mice, the ratio of the injured knee joint’s diameter after injury compared with the baseline was similar to that of the uninjured intra-control knee, which was significantly different compared with the FIXnull control group after injury (Figure 7C). The hemostatic properties in whole blood of 2bCoF9R338L-transduced FIX-primed recipients were further assessed by ROTEM and nWB-TGA. All parameters in both ROTEM and nWB-TGA in 2bCoF9R338L-transduced FIX-primed recipients were significantly improved compared with those in FIXnull controls (Figure 7D-E).
Collectively, these results confirm that platelet-specific 2bCoF9R338L gene therapy can provide therapeutic protein and restore hemostasis once inhibitory antibodies are eradicated in rhFIX-primed FIXnull mice.
Discussion
The central finding of this study is that lentiviral gene delivery of platelet-specific codon-optimized FIX-Padua expression can effectively introduce sustained hyperfunctional FIX expression in platelets in FIXnull mice even with preexisting immunity without anaphylactic reaction. Platelet-targeted hyperfunctional FIX gene therapy can eradicate anti-FIX inhibitors, induce immune tolerance, and provide therapeutic protein once inhibitors drop to undetectable levels. In addition, we found that using bortezomib as a supplemental agent to TBI preconditioning for 2bCoF9R338L gene therapy can significantly accelerate the eradication of anti-FIX inhibitors in primed FIXnull mice.
Our previous studies demonstrated that 2bF9 gene therapy can restore hemostasis and induce immune tolerance in unprimed FIXnull mice.53 However, platelet-FIX:C levels in transduced mice were only ∼3% in whole blood, even when lethal 11-Gy TBI was used.53 In this study, when CoF9R338L was targeted to platelets, platelet-FIX:C levels in transduced unprimed FIXnull mice reached an average level of 35.08 to 60.77 mU/108 platelets, corresponding to around 70% to 122% in whole blood in WT mice. This is a 26.2-fold to 45.4-fold increase compared with our studies using 2bF9LV under a similar transduction and transplantation protocol. Compared with 2bF9 gene therapy, platelet-FIX:Ag levels in 2bCoF9R338L-transduced recipients were 3.6-fold to 4.6-fold higher, which could be attributed to the effect of codon optimization. The platelet-FIX:C/FIX:Ag ratios in 2bCoF9R338L-transduced recipients were comparable to those in reports targeting FIX-Padua expression to the liver in mice,36,58 indicating that platelet-derived vs liver-derived FIX has similar functional activity.
FIX is a vitamin K–dependent protein synthesized by hepatocytes in healthy individuals. It requires posttranslational carboxylation to become biologically active. Because γ-carboxylated glutamic acid residues located in the amino-terminal region can efficiently bind to BaSO4,71 BaSO4 precipitation was used to examine the extent of γ-carboxylation of engineered FIX protein.72 Our studies showed that there was no FIX detected in the supernatants of platelet lysates after BaSO4 precipitation, even in the hyperfunctional FIX-Padua expressers. This confirms that platelet-derived FIX is γ-carboxylated, which demonstrates that megakaryocytes have the capacity to synthesize high levels of hyperfunctional FIX and complete posttranslational carboxylation. The ability of megakaryocytes to synthesize functional, active vitamin K–dependent protein is also evidenced by protein S studies, in which 2.5% of circulating functional protein S is made by megakaryocytes and stored within platelet α-granules.73-75
Our studies show that genetic background could affect FIX neoprotein expression in platelet-targeted gene therapy. In Model 1 (a C57BL/6 background), platelet-FIX expression dropped 75% from week 4 to week 24 after 2bCoF9R338L gene therapy, although the levels of platelet-FIX were maintained afterward (data not shown). In contrast, platelet-FIX expression was sustained after gene therapy in Model 2 (a B6-129S mixed background) in both the noninhibitor and inhibitor model studies. The impact of genetic background on platelet-targeted neoprotein expression is also evidenced in platelet-FVIII gene therapy, in which sustained platelet-FVIII expression was maintained in B6-129S FVIIInull mice,48,52 but it dropped around 54% from week 4 to week 8 in a C57BL/6 background after 2bF8 gene therapy under 6.6-Gy TBI.65 Evidence suggests that genetic background influences both humoral and cellular immune responses.76,77 Because no antibodies against neoprotein were detected in transduced recipients, we speculate that genetic variants between different strains of mice have an impact on the outcome of cellular immune responses in our platelet-specific gene therapy. We know that some CD4 and CD8 T cells are resistant to irradiation, which might lead to an antagonistic process between eliminating the early stages of transduced megakaryocyte progenitors by residual CD8 T cells and inducing immune tolerance by platelet-FIX expression after gene therapy. In the inhibitor model, the platelet-FIX:C levels at the early time points seem to be lower than at later time points, which could be a result of the influence of high-titer inhibitors on the FIX chromogenic assay because any small amount of inhibitory plasma carryover during platelet isolation could neutralize platelet-FIX:C, which would lead to the appearance of lower levels. The other potential reason could be that the FIX-primed immune system has an impact on the uptake of vitamin K, affecting posttranslational carboxylation in the early stages after HSCT.
Gene therapy for HB with preexisting immunity could be challenging because a primed immune system might respond to neoprotein, eliciting a memory immune response or causing anaphylaxis. Furthermore, primed immune cells may kill the transduced cells, leading to gene therapy failure. Our studies show that 2bCoF9R338L gene therapy could prevent anamnestic immune responses and avoid anaphylactic reaction in highly primed FIXnull mice. Using CoF9R338L instead of WT-hFIX significantly improves the functional levels and stability of platelet-FIX expression in platelet-targeted gene therapy in primed FIXnull mice. This could be because 2bCoF9R338L-transduced platelets are more effective than 2bF9-transduced platelets in eradicating or neutralizing anti-FIX inhibitors. Indeed, our results show that there are negative correlations between the levels of platelet-FIX expression and the half-lives of anti-FIX antibody disappearance time in 2bCoF9R338L-transduced recipients.
Although the efficacy of platelet-derived FIX is limited in the presence of anti-FIX inhibitors,54 no bleeding episodes occurred in transduced FIX-primed recipients after platelet-targeted gene therapy. All transduced recipients were well-tolerized to ear-tagging after gene therapy, indicating that platelet-FIX-Padua has at least some functional properties, even in the presence of inhibitors. After inhibitors dropped to undetectable, platelet-FIX-Padua could effectively rescue the bleeding phenotype in FIXnull mice in the tail bleeding test and joint injury. The hemostatic efficacy in primed mice was comparable to that in unprimed mice after 2bCoF9R338L gene therapy, demonstrating that the ultimate goal of gene therapy in providing therapeutic protein was achieved. The efficacy and safety issues of FIX-Padua in AAV-mediated liver-targeted gene therapy have been well documented, with remarkable improvement in hemostasis without increasing thrombosis risk in HB mice, dogs, and humans.33-35,41,58 Our studies show that hemostatic functional properties of platelet-FIX-Padua in FIXnull mice were improved without acceleration of clot formation or thrombin generation compared with WT mice, indicating that platelet-targeted FIX-Padua gene therapy does not increase thrombosis risk in HB mice.
Because the clinical efficacy of platelet-FIX is limited in the presence of inhibitors,54 eliminating antibodies is critical, and a regimen that can delete plasma cells is needed. The proteasome inhibitor bortezomib is a promising candidate because it has been shown to effectively eliminate short- and long-lived plasma cells in other autoimmune disease models67-70,78 and antibodies in patients with HA inhibitors.79 In this study, we showed for the first time that the combination of bortezomib and 6.6 Gy TBI as a preconditioning regimen dramatically decreased anti-FIX inhibitors and antibody disappearance time in primed FIXnull mice, and immune tolerance was established after 2bCoF9R338L gene therapy. Our findings strongly suggest that bortezomib could be used as a supplemental agent in a preconditioning regimen to accelerate the elimination of anti-FIX antibodies for platelet-targeted gene therapy for patients with HB who have preexisting immunity.
In summary, we showed that platelet-specific FIX gene therapy can evade anaphylactic reaction and induce immune tolerance in primed HB mice. Using codon-optimized FIX-Padua remarkably enhances platelet-FIX functional levels and provides long-term therapeutic hyperfunctional FIX protein expression in HB mice, even those with preexisting immunity. A combination of bortezomib with 6.6 Gy TBI preconditioning for platelet FIX gene therapy significantly accelerates the elimination of inhibitors in HB mice. Taken together, platelet-specific 2bCoF9R338L gene therapy could be a promising approach for gene therapy of all HB, even in the high-risk setting of preexisting anti-FIX immunity.
To request data, please send an e-mail to Qizhen Shi at qshi@versiti.org.
Acknowledgments
This work was supported by a grant from the National Institutes of Health, National Heart, Lung, and Blood Institute (HL-102035) (Q.S.), Bayer Hemophilia Foundation Award (Q.S.), and generous gifts from the Children’s Wisconsin Foundation (Q.S.) and Midwest Athletes Against Childhood Cancer Fund (Q.S.).
Authorship
Contribution: J.A.S. designed the study, performed experiments, analyzed data, and edited the manuscript; J.C., Y. Chen, Y. Cai, H.Y., and J.G.M. performed experiments and analyzed data; P.E.M. provided critical materials and edited the manuscript; and Q.S. designed the research, supervised the studies, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Qizhen Shi, Department of Pediatrics, Medical College of Wisconsin, 8701 Watertown Plank Rd, Milwaukee, WI 53226; e-mail: qshi@versiti.org.

