

Key Points
NETosis and caspase-1 activation are tightly interrelated processes, which cooperatively promote DVT.
Pharmacological inhibition of caspase-1 protects against DVT in mice.
Abstract
Deep vein thrombosis (DVT) is linked to local inflammation. A role for both neutrophil extracellular traps (NETs) and the assembly of inflammasomes (leading to caspase-1–dependent interleukin-1β activation) in the development of DVT was recently suggested. However, no link between these 2 processes in the setting of thrombosis has been investigated. Here, we demonstrate that stimulation of neutrophils induced simultaneous formation of NETs and active caspase-1. Caspase-1 was largely associated with NETs, suggesting that secreted active caspase-1 requires NETs as an adhesive surface. NETs and their components, histones, promoted robust caspase-1 activation in platelets with the strongest effect exerted by histones 3/4. Murine DVT thrombi contained active caspase-1, which peaked at 6 hours when compared with 48-hour thrombi. Platelets constituted more than one-half of cells containing active caspase-1 in dissociated thrombi. Using intravital microscopy, we identified colocalized NETs and caspase-1 as well as platelet recruitment at the site of thrombosis. Pharmacological inhibition of caspase-1 strongly reduced DVT in mice, and thrombi that still formed contained no citrullinated histone 3, a marker of NETs. Taken together, these data demonstrate a cross-talk between NETs and inflammasomes both in vitro and in the DVT setting. This may be an important mechanism supporting thrombosis in veins.
Introduction
Deep vein thrombosis (DVT) is a debilitating disease affecting up to 900 000 people annually in the United States.1 Blood flow stagnancy in the veins is one of the leading causes of DVT.2,3 In recent decades, it has been shown that development of DVT is preceded by local inflammation; this process has been termed immunothrombosis.4 In particular, the recruitment of leukocytes and platelets to the venous wall is critical for the onset of thrombosis. Upon stimulation, neutrophils can release neutrophil extracellular traps (NETs), a complex of DNA core, histones, and enzymes,5 which was shown to be involved in thrombosis.6 NETs are a constituent of experimental venous thrombi and may serve as a DVT biomarker in humans.7,8 Genetic inhibition of NET formation or their destruction protects mice from thrombosis.7,9
Neutrophil recruitment can be modulated by interleukin-1β (IL-1β).10-12 Activation and secretion of IL-1β is mediated by inflammasomes. Nod-like receptor protein (NLRP)-3 inflammasomes are molecular complexes primarily focused on the transformation of caspase-1 and caspase-11 into their active forms, which leads to cleavage and activation of IL-1β and IL-18. The link between inflammasomes and DVT has been reported following systemic hypoxia/high altitude.13 In addition, an impact of vascular ecto-apyrase, CD39, in the regulation of IL-1β formation in thrombosis has been demonstrated.14 NETs have been shown to stimulate inflammasome assembly, for example, in the context of systemic lupus erythematosus15 or diabetic wounds.16 In turn, IL-1β stimulates the release of tissue factor associated with NETs.17 However, a relationship between NETs and inflammasomes in different cell types and its role in DVT has not been addressed.
Here, we demonstrate that NETs and inflammasomes in neutrophils and platelets are tightly interrelated and cooperatively support DVT.
Methods
DVT was modeled on C57BL/6 male mice by applying inferior vena cava (IVC) stenosis as described.7 Intravital microscopy was performed 2 hours after DVT surgery; active caspase-1 was detected using FAM-YVAD-FMK FLICA (Bio-Rad), NETs were identified by SYTOX Orange or myeloperoxidase staining, and platelets by an anti-CD41 antibody. All animal experiments were performed in accordance with the UK legislation (Project Licenses 40/3745 and PC427E5DD). Full details of experimental procedures are included in supplemental Methods.
Results and discussion
IL-1β is increased systemically in patients with thrombosis and in animal models of venous thrombosis.18,19 We looked for inflammasome activation in an experimental mouse model of DVT. Activated caspase-1 (FLICA signal) was detected in platelets and leukocytes from dissociated thrombi after 48 hours of IVC stenosis (Figure 1A-B). A small population of platelets in the peripheral blood contained active caspase-1 in the DVT setting (Figure 1A, middle) but the difference in mean fluorescence intensity (MFI) did not reach statistical significance (compared with unchallenged mice; supplemental Figure 1A). Platelets positive for FLICA were activated as judged by the active conformation of integrin αIIbβ3 (supplemental Figure 1B). This corroborates the link between NLRP3 inflammasome assembly and platelet activation demonstrated in vitro.20

Active caspase-1 is present in both thrombi and blood cells in murine DVT and can be induced by histones and NETotic neutrophils in human platelets. (A) FLICA incorporation (active caspase-1) in platelets (from thrombi and blood) and leukocytes 48 hours after IVC stenosis. (B) Quantification of FLICA-positive platelets and leukocytes in both thrombi and blood (n = 5, mean ± standard deviation). (C) Immunofluorescence staining of thrombi sections for FLICA (green), SYTOX Orange (red), and CD41 (blue). Arrows indicate areas of colocalization of FLICA and SYTOX Orange. Scale bars: left = 100 μm, middle = 50 μm, right = 10 μm. (D) Total and cleaved (activated) caspase-1 content in thrombus lysates at either 48 or 6 hours after IVC stenosis (representative gel, n = 4-5). (E) MFI for FLICA incorporation (caspase-1 activation) by washed human platelets stimulated with ATP, nigericin, PAF, and calf histones, at indicated concentrations, or left untreated (n = 3, mean ± standard deviation). One-way analysis of variance, Dunnett’s multiple comparison test, *P ≤ .05. (F) Representative plot of FLICA incorporation by human platelets treated with individual human histones and evaluated by flow cytometry. (G) Immunofluorescent staining of histone-stimulated platelets with FLICA (green) and CD41 (red). Scale bar = 10 µm, n = 3. (H) MFI quantification for FLICA from autologous platelets seeded on NETotic neutrophils after treatment with PMA for 2 hours (n = 3 biological replicates, data points indicate technical replicates). One-way analysis of variance, Tukey’s multiple comparison test, *P ≤ .05.
Active caspase-1 is present in both thrombi and blood cells in murine DVT and can be induced by histones and NETotic neutrophils in human platelets. (A) FLICA incorporation (active caspase-1) in platelets (from thrombi and blood) and leukocytes 48 hours after IVC stenosis. (B) Quantification of FLICA-positive platelets and leukocytes in both thrombi and blood (n = 5, mean ± standard deviation). (C) Immunofluorescence staining of thrombi sections for FLICA (green), SYTOX Orange (red), and CD41 (blue). Arrows indicate areas of colocalization of FLICA and SYTOX Orange. Scale bars: left = 100 μm, middle = 50 μm, right = 10 μm. (D) Total and cleaved (activated) caspase-1 content in thrombus lysates at either 48 or 6 hours after IVC stenosis (representative gel, n = 4-5). (E) MFI for FLICA incorporation (caspase-1 activation) by washed human platelets stimulated with ATP, nigericin, PAF, and calf histones, at indicated concentrations, or left untreated (n = 3, mean ± standard deviation). One-way analysis of variance, Dunnett’s multiple comparison test, *P ≤ .05. (F) Representative plot of FLICA incorporation by human platelets treated with individual human histones and evaluated by flow cytometry. (G) Immunofluorescent staining of histone-stimulated platelets with FLICA (green) and CD41 (red). Scale bar = 10 µm, n = 3. (H) MFI quantification for FLICA from autologous platelets seeded on NETotic neutrophils after treatment with PMA for 2 hours (n = 3 biological replicates, data points indicate technical replicates). One-way analysis of variance, Tukey’s multiple comparison test, *P ≤ .05.
We observed NETs (identified as extracellular DNA by SytoxOrange and myeloperoxidase) in tissue sections of 48-hour thrombi (Figure 1C; supplemental Figure 1C). Active caspase-1 was colocalized with NETs in essentially all fields of view (arrows, Figure 1C). The presence of caspase-1 in thrombi lysates was confirmed by western blotting. Similar quantities of total caspase-1 were detected in thrombi after 6 and 48 hours of IVC stenosis (Figure 1D; supplemental Figure 1D). Processed caspase-1 in thrombi, however, was significantly higher at 6 hours than at 48 hours. Platelets constituted more than one-half of thrombus-derived FLICA-positive cells, suggesting that platelets are an important source of caspase-1 in the DVT setting (supplemental Figure 1E). Taken together, the presence of NETs/caspase-1 complexes and activation of caspase-1 in thrombi imply that these complexes are likely involved in DVT development.
NLRP3 inflammasome has recently been reported in platelets.21-23 We have observed accumulation of NETs/active caspase-1 complexes in venous thrombi at the sites enriched with platelets (Figure 1C) and hypothesized that platelets contribute to inflammasome formation in the setting of DVT. The signal that activates inflammasomes in platelets during thrombosis is unclear, so we screened platelets for potential candidates. Although classical inflammasome activators ATP and nigericin did not induce caspase-1 cleavage, and platelet-activating factor had only a moderate effect, calf histones strongly increased FLICA signal in a dose-dependent fashion (Figure 1E). All individual histones induced caspase-1 activation in platelets with histones 3 and 4 having the strongest effect (Figure 1F; supplemental Figure 2A). Caspase-1 activation developed in parallel with, but independently of, histone-induced platelet aggregation because inhibition of aggregation by Integrilin did not affect FLICA signal (supplemental Figure 2B). The highest aggregation amplitude was induced by histones 3 and 4 (supplemental Figure 2C-D). Stimulation with histone induced expression of activation marker P-selectin in human platelets (supplemental Figure 2E). The pro-aggregatory effect of histones is consistent with previously reported evidence of thrombocytopenia induced by histones in vivo.24 Interestingly, in our experiments, histones induced platelet aggregation in the absence of either fibrinogen or plasma. This may be explained, at least partially, by lower concentrations of individual histones used by the previous studies of Fuchs and colleagues.24 In those studies, 1 μM of H3 and H4 were used, which equals 15.2 and 11.2 μg/mL, respectively, whereas we used 50 μg/mL. Whether histones at higher concentrations form bridges between αIIbβ3 on adjacent platelets independently of fibrinogen or fibrinogen is released from platelet α-granules remains to be elucidated.
NETs are abundantly present in venous thrombi and play a crucial role in DVT.7,25 We therefore next tested whether NETs stimulate inflammasome formation in platelets. Platelets exposed to histone after being seeded on a surface showed bright FLICA signal (Figure 1G). Furthermore, platelets co-incubated with NETotic neutrophils demonstrated increased staining for active caspase-1 (Figure 1H). Thus, histones as components of NETs or NETs as a biological entity promote inflammasome activation in platelets.
We next explored whether NETosis and inflammasome assembly are spatially associated. Both phorbol 12-myristate 13-acetate (PMA) and glucose oxidase (promoting NETosis through activation of protein kinase C and inflicting oxidative damage by hydrogen peroxide, respectively) induced colocalization of active caspase-1 with the speck-like protein containing a caspase-recruitment domain (ASC), suggesting inflammasome assembly, and myeloperoxidase, a marker of NET formation (Figure 2A). About 90% of inflammasome-containing cells were NETotic (Figure 2B), whereas only ∼23% of neutrophils in NETosis also had active caspase-1 (Figure 2C). Thus, NETs may serve as a surface for active caspase-1 accumulation.
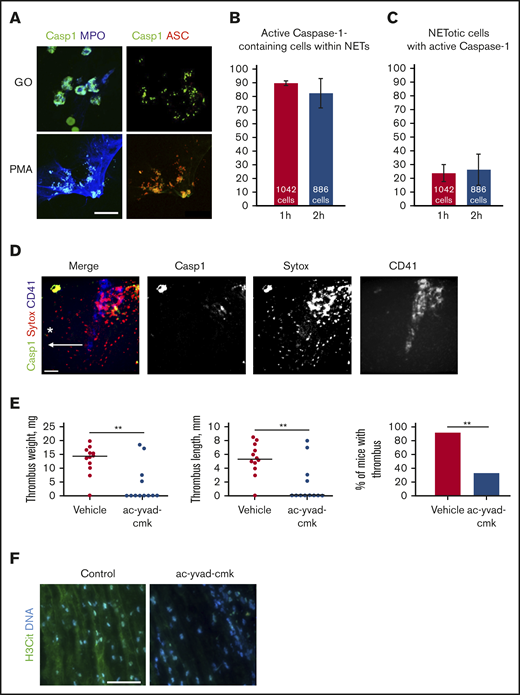
NETs and active caspase-1 colocalize both in vitro and in vivo, and inhibition of caspase-1 reduces DVT. (A) Immunofluorescent staining for activated caspase-1 on NETs formed by human neutrophils stimulated with either PMA or glucose oxidase (FLICA, green; ASC, red; myeloperoxidase, blue). Scale bar = 15 μm. Quantification of the percentage of active caspase-1–containing cells that simultaneously release NETs (B) and the percentage of NETotic cells containing active caspase-1 (C) (n = 3). (D) Z-projection of the murine IVC wall 2 hours after stenosis obtained by intravital microscopy (FLICA/active caspase-1, green; SYTOX Orange, red; CD41, blue). Asterisk depicts the site of stenosis; arrow shows the direction of blood flow. Scale bar = 100 μm, n = 3. (E) Thrombus weight and length, and thrombosis incidence in ac-yvad-cmk (n = 12) and vehicle control-treated mice (n = 12). **P ≤ .01; weight and length, Mann-Whitney U test; incidence, Fisher’s exact test. (F) Citrullinated histone 3 (green) and DNA (blue) immunofluorescence staining of control thrombi and of thrombi developed in mice treated with ac-yvad-cmk. Scale bar = 25 μm, n = 4.
NETs and active caspase-1 colocalize both in vitro and in vivo, and inhibition of caspase-1 reduces DVT. (A) Immunofluorescent staining for activated caspase-1 on NETs formed by human neutrophils stimulated with either PMA or glucose oxidase (FLICA, green; ASC, red; myeloperoxidase, blue). Scale bar = 15 μm. Quantification of the percentage of active caspase-1–containing cells that simultaneously release NETs (B) and the percentage of NETotic cells containing active caspase-1 (C) (n = 3). (D) Z-projection of the murine IVC wall 2 hours after stenosis obtained by intravital microscopy (FLICA/active caspase-1, green; SYTOX Orange, red; CD41, blue). Asterisk depicts the site of stenosis; arrow shows the direction of blood flow. Scale bar = 100 μm, n = 3. (E) Thrombus weight and length, and thrombosis incidence in ac-yvad-cmk (n = 12) and vehicle control-treated mice (n = 12). **P ≤ .01; weight and length, Mann-Whitney U test; incidence, Fisher’s exact test. (F) Citrullinated histone 3 (green) and DNA (blue) immunofluorescence staining of control thrombi and of thrombi developed in mice treated with ac-yvad-cmk. Scale bar = 25 μm, n = 4.
Because we had observed an association between NETosis and caspase-1 activation in vitro and ex vivo, we next addressed whether they interact also in in vivo DVT conditions. Intravital imaging of the IVC near the ligation site at the time point preceding thrombus formation (2 hours after stenosis application) further confirmed colocalization of NETs with activated caspase-1 (Figure 2D). NETs occupied much larger areas than active caspase-1 and were diffuse rather than associated with cell-like structures.
Because NET destruction prevents thrombosis,7 we hypothesized that caspase-1 could be a potential target in DVT, which is supported by recent evidence of the protective effect of targeting IL-1β or its receptor.14 Indeed, administration of a selective caspase-1 inhibitor reduced thrombosis in mice (Figure 2E). The caspase-1 inhibitor has been used in vivo in multiple studies, and its effect is similar to hereditary caspase-1 deficiency,26 which minimizes the possibility of its off-target effects. A small proportion of the inhibitor-treated animals still formed thrombi of regular size (Figure 2E). These residual thrombi from mice administered with caspase-1 inhibitor contained very low levels of citrullinated histone 3 (Figure 2F), a marker of NETs abundantly present in thrombi from control animals. This suggests existence of biological feedback, through which suppression of caspase-1 leads to downregulation of NETosis in the DVT setting. The biological effects of caspase-1 inhibition may not be identical to neutralization of IL-1β as multiple downstream effects of this cytokine occur independently of caspase-1 with IL-1β being activated by other enzymes.27,28 Caspase-1 is directly implicated in blood coagulation induced by Escherichia coli T3SS inner rod protein EprJ as judged by prothrombin time, plasma fibrinogen concentration, and TAT complexes.29 This mechanism could be implicated in the DVT setting as well. Thus, targeting caspase-1 for DVT prevention might be safer and produce less side effects such as, for example, fatal infection and sepsis, observed in the Canakinumab Anti-inflammatory Thrombosis Outcomes Study.30 Drugs inhibiting caspase-1 activity are currently under development.31,32
In conclusion, we have demonstrated an association between formation of NETs and inflammasome assembly both in vitro and in DVT (supplemental Figure 3).
Send data sharing requests via e-mail to the corresponding author, Alexander Brill (a.brill@bham.ac.uk).
Acknowledgments
This work was supported by a British Heart Foundation Project Grant (PG/18/46/33817) (A.B.). A.B. is supported by a British Heart Foundation Senior Basic Science Research Fellowship (FS/19/30/34173). A.O.K. is supported by a Henry Wellcome Fellowship (218649/Z/19/Z).
Authorship
Contribution: A.B., T.P., and J.C. were responsible for study concept and design; T.P., J.C., A.D.P., K.W., C.W.S., A.O.K., and D.K. acquired the data; A.B., T.P., A.D.P., D.K., and J.C. analyzed and interpreted the results; A.B. supervised the work and obtained funding; A.B. and J.C. wrote the manuscript; and A.B., T.P., A.D.P., D.K., and J.C. critically revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for T.P. is Plasticell Ltd, Stevenage, United Kingdom.
The current affiliation for A.D.P. is Diana Princess of Wales Hospital, Northern Lincolnshire and Goole NHS Foundation Trust, Grimsby, United Kingdom.
Correspondence: Alexander Brill, Institute of Cardiovascular Sciences, College of Medical and Dental Sciences, University of Birmingham, Edgbaston, Birmingham B15 2TT, United Kingdom; e-mail: a.brill@bham.ac.uk.

