

Key Points
An approach called CRIMSON uses CRISPR-edited megakaryocytes for rapid assessment of genes associated with platelet function.
CRIMSON defines platelet-like responses in megakaryocytes after deleting platelet genes ITGA2B, RASGRP2, GP6, B2M, and novel gene COMMD7.

Abstract
Human anucleate platelets cannot be directly modified using traditional genetic approaches. Instead, studies of platelet gene function depend on alternative models. Megakaryocytes (the nucleated precursor to platelets) are the nearest cell to platelets in origin, structure, and function. However, achieving consistent genetic modifications in primary megakaryocytes has been challenging, and the functional effects of induced gene deletions on human megakaryocytes for even well-characterized platelet genes (eg, ITGA2B) are unknown. Here we present a rapid and systematic approach to screen genes for platelet functions in CD34+ cell-derived megakaryocytes called CRIMSON (CRISPR-edited megakaryocytes for rapid screening of platelet gene functions). By using CRISPR/Cas9, we achieved efficient nonviral gene editing of a panel of platelet genes in megakaryocytes without compromising megakaryopoiesis. Gene editing induced loss of protein in up to 95% of cells for platelet function genes GP6, RASGRP2, and ITGA2B; for the immune receptor component B2M; and for COMMD7, which was previously associated with cardiovascular disease and platelet function. Gene deletions affected several select responses to platelet agonists in megakaryocytes in a manner largely consistent with those expected for platelets. Deletion of B2M did not significantly affect platelet-like responses, whereas deletion of ITGA2B abolished agonist-induced integrin activation and spreading on fibrinogen without affecting the translocation of P-selectin. Deletion of GP6 abrogated responses to collagen receptor agonists but not thrombin. Deletion of RASGRP2 impaired functional responses to adenosine 5′-diphosphate (ADP), thrombin, and collagen receptor agonists. Deletion of COMMD7 significantly impaired multiple responses to platelet agonists. Together, our data recommend CRIMSON for rapid evaluation of platelet gene phenotype associations.
Introduction
Numerous genes implicated in platelet function have been identified through candidate gene studies, genome-wide association studies, rare variant association analysis, gene expression association, and exome and genome sequencing of patients and families with platelet disorders. However, genetic association studies require additional evidence to establish causation. Unfortunately, genetic modifications in human platelets have been difficult because of their size, lack of nuclear DNA, and the difficulty of maintaining them in a quiescent state for long periods. Therefore, studies of platelet gene function have traditionally relied on mouse models and alternative human cell models. Mouse studies have yielded significant insights into platelet gene functions, but they are expensive and slow, which renders them impractical for screening studies. Moreover, some orthologs for human genes are not expressed or diverge considerably in mice (we have shown that ∼17% differ more than 100-fold in expression),1 and gene functions in mice may not fully reflect those of their human counterparts.2 This was exemplified by Coughlin et al3 when they observed no platelet phenotype after knockout (KO) of coagulation factor II thrombin receptor (PAR1) in mice.
Megakaryocyte (MK)-like cells and platelet-like particles harvested from them are increasingly used as models for human platelet studies. Cell lines such as MEG-01,4 and more recently immortalized megakaryocyte progenitor cell lines (imMKCLs),5 offer replenishable sources of MK-like cells that can be genetically manipulated. Although they provide useful and complementary information, studies in cell lines have known limitations, including lack of genetic diversity. Alternatively, primary MK models, which include induced pluripotent stem cells (iPSCs) and CD34+ hematopoietic progenitor–derived cells, reflect human genetic diversity and are the closest nucleated cells to platelets in origin, structure, and function. Unlike iPSCs, which take significant time and labor to generate, we and others have shown that mature MK-like cells can be derived from CD34+ hematopoietic progenitors in just 2 weeks.6 Notwithstanding inevitable differences between circulating platelets and cultured megakaryocytes (see “Discussion” for limitations), cumulative evidence indicates that CD34+-derived MK-like cells (hereafter referred to as MKs) partially mimic select classic platelet functional responses. They adhere to and spread on collagen and fibrinogen, they are known to aggregate, and they activate integrins and release granules in response to platelet agonists such as thrombin, collagen-related peptide (CRP), and adenosine 5′-diphosphate (ADP).7-15
Until recently, gene editing of CD34+-derived primary MKs has been challenging and inefficient, or it relied mostly on the use of viral transduction (ie, lentivirus) with sorting or drug selection.16 Recent studies have overcome this limitation by using clustered regularly interspaced short palindromic repeats (CRISPR) with CRISPR-associated protein 9 (Cas9) ribonucleoprotein (RNP) transfection to achieve highly efficient, nonviral gene editing in CD34+ cells.17 Two published studies have further differentiated RNP/CRISPR-edited CD34+ cells into megakaryocytes.18,19 However, these 2 studies focused on single-gene targets (ETV6 and LCP1) to interrogate megakaryopoiesis without providing information on MK yield, purity, or viability. Furthermore, previous studies did not assess classic platelet functions. Thus, we do not know the extent to which primary MK functional responses recapitulate expected human platelet responses after gene editing. This is the case even for well-characterized platelet genes such as ITGA2B, GP6, or RASGRP2.
Here we demonstrate a rapid, nonviral, highly efficient approach using CRISPR/Cas9 to obtain gene-edited MKs, and we use these to interrogate classic platelet gene functions and overcome current limitations in the field. We term our approach CRIMSON (CRISPR-edited megakaryocytes for rapid screening of platelet gene functions). We use CRIMSON to delete 3 critical platelet genes known for their involvement in platelet activation responses (GP6, RASGRP2, and ITGA2B) and evaluate the effect of each gene deletion on a select set of platelet-like functional responses in MKs. For comparison, we delete the immune receptor component B2M. Finally, we use CRIMSON to assess whether COMMD7, a gene previously associated with cardiovascular disease and platelet function, mediates responses to platelet agonists.
Materials and methods
CD34+-derived primary MK differentiation
The study was approved by the University of Utah Institutional Review Board and was conducted in accordance with the Declaration of Helsinki. Cord blood was obtained from the New York Blood Bank and Cleveland Cord Blood Center and was used in accordance with approval by the University of Utah Institutional Review Board. CD34+ hematopoietic progenitors were isolated via positive selection with magnetic microbeads (Miltenyi, #130-046-702) and differentiated into megakaryocytes as previously described.6 Briefly, isolated cells were plated in 12-well plates at 5 × 105 cells per mL in Serum-Free Expansion Medium (SFEM; Stem Cell Technologies, #09650) supplemented with thrombopoietin (TPO, 20 ng/mL; Peprotech, #300-18), and stem cell factor (25 ng/mL; Peprotech, #300-07) until day 6 and SFEM plus TPO (50 ng/mL) thereafter. Cells were passaged into fresh media back to 1 × 106 cells per mL on days 3, 6, and 10, and analyzed on day 13.
CRISPR/Cas9 transfection
CD34+ cells were transfected with CRISPR/Cas9 complexes on day 5 (between day 0 and day 9 for optimization experiments) of culture using minor modifications of published protocols17,20 to accommodate our MK culture conditions, which were recently published.18 Predesigned Alt-R CRISPR/Cas9 CRISPR RNAs (crRNA) with >65% on-target score were purchased from Integrated DNA Technologies (IDT, Coralville, IA). 1 to 2 crRNAs with the highest cutting efficiency were chosen from up to 4 crRNAs screened for each target. The IDT company ID (annotated by double letters) and sequences of the crRNAs used were ITGA2B AA (called sequence A in the text): CAGAGTACTTCGACGGCTAC, AF (sequence B [selected for highest efficiency]): GGACAAGCGTTACTGTGAAG, AO (sequence C): GGGAGGACACGTGCCACAAA; GP6 AL: CATACCGAGTCTGACACTGT, RASGRP2 AJ: CCTGGACAAGGGCTGCACGG, B2M AA: CGTGAGTAAACCTGAATCTT, B2M AB: AAGTCAACTTCAATGTCGGA, COMMD7 AA: ACCTATGGCCCATCGAGCAA, and COMMD7 AC: ACCACCAATCAGATCAGTCT. IDT negative control #1 was used as a nontargeting control. All materials and reagents were kept sterile and RNAse free throughout. crRNA, trans-activating CRISPR RNA (tracrRNA) (IDT #1072532), and electroporation enhancer (IDT #1075915) were resuspended to 100 µM in 10 mM tris(hydroxymethyl)aminomethane (Tris; pH 7.5), and stored at −20°C until use. crRNA and tracRNA were mixed 1:1 (2.5 µL each) in a polymerase chain reaction (PCR) tube and heated at 95°C for 5 minutes in a thermal cycler with a heated lid, followed by cooling to room temperature on a bench top for at least 10 minutes before storing at −20°C.
Immediately before transfection, RNP complexes were formed by mixing (gentle swirling with pipet tip) the following in a PCR tube: for 2 cords, 2.1 µL of phosphate-buffered saline (PBS; no calcium or magnesium), 1.2 µL of crRNA-tracRNA duplex, and 1.7 µL Alt-R Streptococcus pyogenes (S.p.) (Alt-R S.p.) Cas9 V3 (62 µM stock, IDT #1081058). The mixture was heated in a thermal cycler at 37°C for 4 minutes and allowed to cool to room temperature on a bench top for 10 minutes. Simultaneously, cells were counted, washed once with PBS (centrifuged at 200g at room temperature), and suspended at 0.25 × 106 to 4 × 106 cells per transfection in a PCR tube containing 20 µL of room temperature Amaxa nucleofector solution P3 (Lonza, Basel, Switzerland). Then, 2.5 µL of RNP complex (for double CRISPR cotransfections, use 2.5 µL of each) was gently mixed with 20 µL of cells and 1 µL of enhancer and transferred to a 16-well electroporation strip. Cells were transfected using Amaxa nucleofector 4D program DZ100. Immediately following transfection, 75 µL of prewarmed media (SFEM with TPO-stem cell factor) was added to the cuvette, left to sit for 5 minutes at room temperature, then gently transferred to a 24-well plate containing 500 µL of media. Finally, 24 hours later, cells were passaged and culture was continued as described above.
MK activation and analysis
MK analyses were performed on day 13 using bulk culture MKs. For DNA analysis, DNA was extracted, the region surrounding CRISPR cut sites was amplified by PCR, and amplicons were sequenced with Sanger sequencing. For protein analysis, cells were snap frozen and lysed with NP40 lysis buffer followed by Laemmli sample buffer; phospho-proteins were directly lysed with Laemmli sample buffer. Denatured, reduced proteins were separated on 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and were transferred to polyvinylidene difluoride membranes. Proteins were detected with antibodies, including calcium diacylglycerol-guanine exchange factor 1 (CalDAG-GEF1) (RASGRP2) (Genetex #GTX108616), αIIb (Santa Cruz #166599), GP6 (R&D Systems #AF3627), B2M (Abcam #Ab75853), COMMD7 (Novus Biologicals #NBP2-58399), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Santa Cruz # 47724), and phospho-protein kinase C (pPKC) substrates (Cell Signaling #6967). This was followed by horseradish peroxidase–conjugated secondary antibodies and chemiluminescent detection. Active RAP1-guanosine-5′-triphosphate (GTP) was detected by using a RAP1 activity assay kit (EMD Millipore, #17-321). For COMMD7 RNA expression analysis, MKs were lysed in and total RNA was isolated from Trizol followed by treatment with DNAse and reverse transcription with superscript IV (Invitrogen). COMMD7 was amplified using primers 5′-CAGTCTTGGCTCCCTCAGAA-3′ and 5′-CAGAGTCTGACCTATGGCCC-3′ and detected in real time using SYBR green dye. Amplicons were sequenced to verify that the correct target was amplified. Expression was normalized to GAPDH, and relative expression was calculated using the 2-ΔΔCT method.
For differentiation marker analysis, MKs were stained with fluorophore-conjugated antibodies against CD41, CD61, CD42a, or immunoglobulin controls for 15 minutes at room temperature. For activation analysis, cells were treated for 15 minutes at 37°C with 1 U/mL thrombin, 2 µg/mL cross-linked CRP (CRP-XL), 2.5 µg/mL convulxin, or 200 ng/mL 2-methylthio-adenosine-5′-diphosphate (2-MeSADP) in 100 µL at a concentration of 106 cells per mL in SFEM media in the presence of PAC1-fluorescein isothiocyanate, P-selectin-phycoerythrin, CD63 V450 antibodies (BD Biosciences, #340507, #555524, #561984) or Alex-Fluor 647–labeled fibrinogen from human plasma (Invitrogen, #F35200). Cells were fixed with 2% paraformaldehyde and analyzed on a Cytoflex flow cytometer (Beckman Coulter). The mean fluorescent intensity (MFI) or percent of cells bound to PAC-1, P-selectin, or fibrinogen was normalized among different batches of cords by taking the percent activated compared with each agonist’s negative control CRISPR split from the same cord, followed by normalization to the mean across all cords.
MK numbers, morphology, and spreading
Cells were stained with Trypan Blue, and viability and counts were assessed by using the TC-20 automated cell counter (Biorad). For spreading, MKs were plated on collagen- (type I; Chrono-log #385) or fibrinogen- (from human plasma; EMD-Millipore #341576) treated 96-well plates for 1 hour. Unbound cells were removed by a gentle wash with PBS, and cells were fixed with 2% paraformaldehyde. Cells were permeabilized with 0.1% Triton × 100 and stained with phalloidin 488; morphology was visualized using an Evos microscope. The ratio of spread to unspread cells was scored blinded to conditions.
Statistics
Two-group significance testing was performed using paired Student t test. A mixed effects model was used for multigroup significance testing followed by Dunnett’s adjustment for multiple comparisons in which each group was compared with the negative control group. P < .05 was considered significant. However, as additional information, P values between < .1 and > .05 are reported for each figure, and P values < .05 are reported as *P < .05, **P < .01, ***P < .001, ****P < .0001. The significance test used and sample sizes are reported in each figure legend.
Results
Rapid and efficient gene deletion using CRISPR in MKs
Mediators of platelet function were genetically deleted in MKs by using nonviral delivery of CRISPR/Cas9 RNP complexes into CD34+ cells according to the methods outlined in Figure 1A and described in the “Materials and methods” section. By using this method, we first screened 3 different crRNAs (called CRISPR-A, -B, and -C) predicted to target ITGA2B (which codes the αIIb subunit of the platelet fibrinogen receptor) for their effect on αIIb (CD41) surface expression. As shown in supplemental Figure 1A, a significant loss of surface αIIb was observed for 2 of 3 crRNAs, with complete ablation in 94% of cells for the CRISPR-B crRNA, which was selected for the remainder of the experiments. By using this crRNA, we tested additional parameters of MK CRISPR editing. As indicated in supplemental Figure 1B, efficient editing using single CRISPRs in MKs was achieved with electroporation of CRISPR/Cas9 into between 0.5 × 106 and 4 × 106 CD34+ cells. Notably, efficient CRISPR editing could be consistently and reproducibly performed up to day 6 of MK culture (Figure 1B). On the basis of the above results, transfection of 0.5 × 106 to 1 × 106 cells on day 5 was selected for the remainder of the experiments.
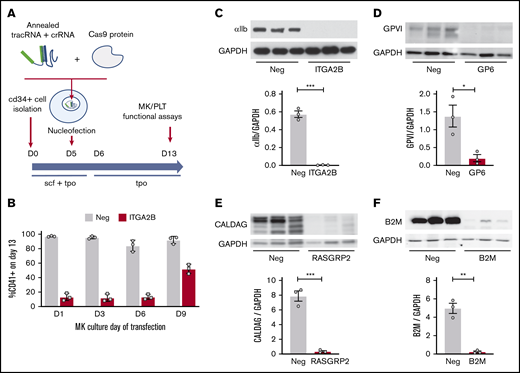
Efficient CRISPR/Cas9 deletions of platelet function genes are maintained in CD34+-derived MKs. (A) Overview of strategy. CD34+ cells isolated from cord blood were transfected on day 5 (D5) (unless otherwise noted) with RNP complexes containing tracrRNA, gene targeting crRNA, and Cas9 protein. MK assays, including assays normally used to measure platelet (PLT) functional responses, were performed on day 13 of culture. (B) CD34+ cells were transfected on different days of culture with negative (Neg) control or ITGA2B crRNA. Surface expression of αIIb (% CD41+ cells; see Figure 2B for gating) was measured by flow cytometry on day 13 MKs. (C-F) Western blot and densitometry analysis of proteins on day 13 MKs after targeting by negative control or the indicated gene specific CRISPR on day 5. Data are presented as mean ± standard error of the mean (SEM) (3 independent cords per group). Unpaired Student t test: *P < .05; **P < .01; ***P < .001. CalDAG, calcium diacylglycerol; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Efficient CRISPR/Cas9 deletions of platelet function genes are maintained in CD34+-derived MKs. (A) Overview of strategy. CD34+ cells isolated from cord blood were transfected on day 5 (D5) (unless otherwise noted) with RNP complexes containing tracrRNA, gene targeting crRNA, and Cas9 protein. MK assays, including assays normally used to measure platelet (PLT) functional responses, were performed on day 13 of culture. (B) CD34+ cells were transfected on different days of culture with negative (Neg) control or ITGA2B crRNA. Surface expression of αIIb (% CD41+ cells; see Figure 2B for gating) was measured by flow cytometry on day 13 MKs. (C-F) Western blot and densitometry analysis of proteins on day 13 MKs after targeting by negative control or the indicated gene specific CRISPR on day 5. Data are presented as mean ± standard error of the mean (SEM) (3 independent cords per group). Unpaired Student t test: *P < .05; **P < .01; ***P < .001. CalDAG, calcium diacylglycerol; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Efficient CRISPR/Cas9 cutting should lead to insertions and deletions (editing) at the DNA cut site, which can cause loss of gene expression. Thus, we amplified DNA from day 13 MKs and sequenced the surrounding CRISPR target site to directly verify maintenance of CRISPR cutting as MKs become polyploid. As shown in supplemental Figure 1C-D, there was a high level of DNA editing for ITGA2B crRNA, indicating that CRISPR DNA edits generated on day 5 of culture are maintained as MKs mature to day 13 of culture.
CRISPR targeting of multiple platelet function genes in MKs
We expanded CRISPR targets to include GP6 (glycoprotein VI [GPVI] platelet collagen receptor), RASGRP2 (the calcium intracellular signaling molecule CalDAG-GEF1), and B2M (the major histocompatibility complex I [MHC I] β2 microglobulin subunit). B2M was chosen as an abundantly expressed control target that is not known to regulate platelet activation responses but that has high relevance to immunology and transfusion medicine. As shown in Figure 1C-E, single crRNA cutting resulted in nearly complete loss in total protein expression for ITGA2B, GP6, and RASGRP2 (mean ± standard deviation [for this paragraph]: 99% ± 0.4%, 87% ± 6%, 95% ± 2%). B2M protein was reduced to a lesser extent (66% ± 15%) using single crRNA (data not shown). This was improved to 95% ± 3% when 2 different crRNAs (B2M crRNA AA and AB) were combined (Figure 1F; supplemental 1F). For the surface proteins αIIb, GPVI, and B2M, loss of total protein coincided with loss of surface expression (supplemental Figure 1E-F). Because CRISPR DNA editing resulted in deletion or KO of protein, we hereafter use the terms CRISPR editing, deletion, and KO interchangeably.
Maturation of MKs after CRISPR KO
Following a 1-day recovery after transfection on day 5, KO and control cells maintained similar viability between days 6 and 13 of culture (Figure 2A) and expanded ∼fivefold (5.1 ± 2.6) between days 6 and 13 (supplemental Figure 2). Control and KO cultures from all groups contained large polyploid cells characteristic of mature MKs (supplemental Figure 3). Flow cytometry analysis of CRISPR KO cells with markers of MK maturation, and as gated in Figure 2B, indicated specific target gene reduction with no off-target reduction in other markers of maturation (Figure 2C-F). For example, in the representative flow cytometry dot plots in Figure 2C (top right panel), surface expression of CD42a (GPIX) is maintained even when surface expression of αIIb is ablated by CRISPR targeting. However, integrin β3 (CD61), the heterodimer partner protein for αIIb (CD41), was also significantly diminished (Figure 2C, bottom right panel) in ITGA2B crRNA targeted cells, as expected. Otherwise, MK maturation markers CD41, CD61, and CD42a were maintained after deletion of GP6, RASGRP2, and B2M (Figure 2C-F). Thus, our approach to ablate proteins in MKs does not seem to have off-target effects for these specific targets on MK proliferation or maturation.
![CRISPR KO cells differentiate into MKs. (A) Percent viable negative control and KO cells on days 6 to 13 of culture after CRISPR/Cas9 on day 5 (3 independent cords per group). (B) Representative flow cytometry gating strategy for MKs on day 13 of culture: single MKs were first gated on larger, lower granularity cells according to forward scatter (FSC-A) and side scatter (SSC-A) as previously published6 and further gated against doublets (FSC-H [height] vs FSC-W [width]). (C) Representative flow cytometry analysis of MK maturation markers on negative control and ITGA2B CRISPR MKs. MKs were gated as in panel B, and positive gates for MK maturation markers CD41 (x-axis), CD42a (y-axis, top panels), or CD61 (y-axis, bottom panels) were set with reference to isotype (immunoglobulin [Ig]) controls. (D-F) Mean ± SEM of the percent of cells expressing MK markers (y-axis) after deletion with CRISPRs targeting the genes listed on the x-axis (3 independent cords per group). Mixed effects analysis with Dunnett’s adjustment for multiple comparisons. Data are presented as mean ± SEM. Paired Student t tests: **P < .01; ****P < .0001.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/9/10.1182_bloodadvances.2020004112/1/m_advancesadv2020004112f2.png?Expires=1769088844&Signature=T43tS9~UGfcjSkPFmbiwmq9X8tQnYs714LXkJSZ1j17BDDM~a7YovNepLH7tCeNa3YLELm8ndXs571uydPH7P1xJZE30Imk6Vi46no0e-QKpqueHCbzJ1zv2uqLwSxSI2zePY~oDYZzHXjSlSYqlflFqFA4rxlYSo~O0k2Iv-00tf~nbPK8CKBZ9zzJ-hzOrdaPEhzasN9wavTR-oYwBibwBmJXThdcaAeIdwGk1iaPfwxfT4TT8~7HFCyrViyzmzL1NX~0oLFsNGAMCJmEb4-czkvqJG8f8Or2frTVfQLIglMLkBgFbj4zFwLnx2yToFn8Fss4OLHAK3CJcGqJWQg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CRISPR KO cells differentiate into MKs. (A) Percent viable negative control and KO cells on days 6 to 13 of culture after CRISPR/Cas9 on day 5 (3 independent cords per group). (B) Representative flow cytometry gating strategy for MKs on day 13 of culture: single MKs were first gated on larger, lower granularity cells according to forward scatter (FSC-A) and side scatter (SSC-A) as previously published6 and further gated against doublets (FSC-H [height] vs FSC-W [width]). (C) Representative flow cytometry analysis of MK maturation markers on negative control and ITGA2B CRISPR MKs. MKs were gated as in panel B, and positive gates for MK maturation markers CD41 (x-axis), CD42a (y-axis, top panels), or CD61 (y-axis, bottom panels) were set with reference to isotype (immunoglobulin [Ig]) controls. (D-F) Mean ± SEM of the percent of cells expressing MK markers (y-axis) after deletion with CRISPRs targeting the genes listed on the x-axis (3 independent cords per group). Mixed effects analysis with Dunnett’s adjustment for multiple comparisons. Data are presented as mean ± SEM. Paired Student t tests: **P < .01; ****P < .0001.
CRISPR KO cells differentiate into MKs. (A) Percent viable negative control and KO cells on days 6 to 13 of culture after CRISPR/Cas9 on day 5 (3 independent cords per group). (B) Representative flow cytometry gating strategy for MKs on day 13 of culture: single MKs were first gated on larger, lower granularity cells according to forward scatter (FSC-A) and side scatter (SSC-A) as previously published6 and further gated against doublets (FSC-H [height] vs FSC-W [width]). (C) Representative flow cytometry analysis of MK maturation markers on negative control and ITGA2B CRISPR MKs. MKs were gated as in panel B, and positive gates for MK maturation markers CD41 (x-axis), CD42a (y-axis, top panels), or CD61 (y-axis, bottom panels) were set with reference to isotype (immunoglobulin [Ig]) controls. (D-F) Mean ± SEM of the percent of cells expressing MK markers (y-axis) after deletion with CRISPRs targeting the genes listed on the x-axis (3 independent cords per group). Mixed effects analysis with Dunnett’s adjustment for multiple comparisons. Data are presented as mean ± SEM. Paired Student t tests: **P < .01; ****P < .0001.
Functional assessment after CRISPR KO in MKs
We next assessed several classic platelet functional responses in the context of CRISPR deletions including signaling, integrin activation, P-selectin exposure, substrate binding, and spreading. Because multiple activation signaling pathways in platelets converge on PKC, we tested the effect of GPVI KO on PKC activity in MKs. As shown in supplemental Figure 4, convulxin or thrombin induced PKC activity in control megakaryocytes, but only thrombin and not convulxin induced PKC activity in GPVI-deficient megakaryocytes, which confirms that GPVI is specifically required to transduce this signal in MKs.
Integrin activation and degranulation of MKs was assessed at baseline and in response to 10 minutes of treatment with the platelet agonists thrombin, CRP, or ADP using flow cytometry analysis of classic markers of platelet activation: PAC-1 binding (activated αIIb/β3) and P-selectin surface exposure (a marker of degranulation). Representative flow cytometry plots of P-selectin and PAC-1 staining on unstimulated or thrombin-stimulated control or ITGA2B KO MKs are shown in Figure 3A. In this example, unstimulated MKs had low levels of activated αIIb/β3 (<1% of cells) and surface P-selectin (3.5%). Thrombin treatment (1 U/mL) resulted in 68% of MKs being stained positive for PAC-1 and 70% of MKs being stained positive for P-selectin. However, when ITGA2B was deleted in MKs from the same cord, PAC-1 binding was reduced to <1% without a reduction in the expression of surface P-selectin. This demonstrative pattern is similar to that observed for platelets from patients with Glanzmann thrombasthenia,21 and it highlights the specificity and potential utility of the approach.
![Integrin activation and P-selectin exposure of CRISPR KO MKs in response to platelet agonists. Day 13 CRISPR (CR) KO and negative control CRISPR MKs were unstimulated (baseline [BL]) or treated for 10 minutes with thrombin (THR) (1 U/mL), CRP (1 μg/mL), or 2-methylthio-adenosine-5′-diphosphate (2-MeSADP; 200 nM). They were then stained for activated αIIb/β3 (PAC-1) and P-selectin and were analyzed by flow cytometry. (A) Representative flow cytometry plots of unstimulated (Unstim) vs thrombin-stimulated negative control and ITGA2B KO MKs. MKs were first gated as described in the legend for Figure 2B. Positive gates for PAC-1 (x-axis) and P-selectin (y-axis) were then set in reference to isotype (Ig) controls. (B-C) Normalized mean ± SEM summaries of the percentage of PAC-1–positive (B) or P-selectin–positive (C) cells (3-9 independent cords per group) for the CRISPR KO indicated on the x-axis. Mixed effects analysis with Dunnett’s adjustment for multiple comparisons: *P < .05; ***P < .001; ****P < .0001.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/9/10.1182_bloodadvances.2020004112/1/m_advancesadv2020004112f3.png?Expires=1769088844&Signature=yGPlCfVLGA~vd7M-YOgwPnWkjnXZd4l7mJB-35Lfttg6CvsT7NHRQi0topSva6yN~7iFLlQsWQFRoZqk5Hr7sqA9uhQQ1daCVwhkIZQbMUQn1OGk171S8UTFzfZ8y-OYcZB-X3asnrtsqYp-SwM8wslvvTrqKsDKgaNsr6f6C4pBqJ72cRRXUoocOlIe3n-3VxVdD8K2EzPlso98dTpkGzbUDBL1PrbtaiPtsiL~UP-7DTrBVVYKApojm67BzPlLnp8R1--kfGueGC5hzbNUOvjv4jgxVHg-73vutoLDJHhRNPGKGX3xOD5-5vW2ac~MkryIRW8FQYQfOdIVek7Umw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Integrin activation and P-selectin exposure of CRISPR KO MKs in response to platelet agonists. Day 13 CRISPR (CR) KO and negative control CRISPR MKs were unstimulated (baseline [BL]) or treated for 10 minutes with thrombin (THR) (1 U/mL), CRP (1 μg/mL), or 2-methylthio-adenosine-5′-diphosphate (2-MeSADP; 200 nM). They were then stained for activated αIIb/β3 (PAC-1) and P-selectin and were analyzed by flow cytometry. (A) Representative flow cytometry plots of unstimulated (Unstim) vs thrombin-stimulated negative control and ITGA2B KO MKs. MKs were first gated as described in the legend for Figure 2B. Positive gates for PAC-1 (x-axis) and P-selectin (y-axis) were then set in reference to isotype (Ig) controls. (B-C) Normalized mean ± SEM summaries of the percentage of PAC-1–positive (B) or P-selectin–positive (C) cells (3-9 independent cords per group) for the CRISPR KO indicated on the x-axis. Mixed effects analysis with Dunnett’s adjustment for multiple comparisons: *P < .05; ***P < .001; ****P < .0001.
Integrin activation and P-selectin exposure of CRISPR KO MKs in response to platelet agonists. Day 13 CRISPR (CR) KO and negative control CRISPR MKs were unstimulated (baseline [BL]) or treated for 10 minutes with thrombin (THR) (1 U/mL), CRP (1 μg/mL), or 2-methylthio-adenosine-5′-diphosphate (2-MeSADP; 200 nM). They were then stained for activated αIIb/β3 (PAC-1) and P-selectin and were analyzed by flow cytometry. (A) Representative flow cytometry plots of unstimulated (Unstim) vs thrombin-stimulated negative control and ITGA2B KO MKs. MKs were first gated as described in the legend for Figure 2B. Positive gates for PAC-1 (x-axis) and P-selectin (y-axis) were then set in reference to isotype (Ig) controls. (B-C) Normalized mean ± SEM summaries of the percentage of PAC-1–positive (B) or P-selectin–positive (C) cells (3-9 independent cords per group) for the CRISPR KO indicated on the x-axis. Mixed effects analysis with Dunnett’s adjustment for multiple comparisons: *P < .05; ***P < .001; ****P < .0001.
Each agonist and CRISPR combination followed the same pattern (but with some added insights) of mimicking expected platelet responses that have been learned from studies of patients with the corresponding genetic deficiencies (Figure 3B-C; for MFIs, see supplemental Figure 5). Thrombin, CRP, and ADP positively induced αIIb/β3 activation to a similar level in negative control compared with B2M KO MKs, although ADP responses were weak with only a small fraction of MKs responding (Figure 3B). In contrast, none of the agonists induced αIIb/β3 activation in ITGA2B KO MKs.21 GPVI KO abolished αIIb/β3 activation in response to CRP, and not thrombin, as expected,22,23 but surprisingly it also impaired responses to ADP. RASGRP2 KO abolished ADP-mediated αIIb/β3 activation and impaired its activation in response to CRP. This is consistent with the known role of RASGRP2 as a requisite intermediate in ADP-induced signaling and a nonrequisite participant in CRP-induced signaling.24,25
Thrombin and CRP triggered robust P-selectin exposure in negative control and B2M KO MKs (Figure 3C; supplemental Figure 5B shows the same data as MFI), which reached similar levels in RASGRP2 and ITGA2B KO MKs. In contrast, GP6 KO abolished P-selectin exposure after treatment with CRP but not thrombin treatment. In platelets, ADP exposure generally elicits weak α-granule degranulation (P-selectin exposure) compared with integrin activation or responses to thrombin or GPVI agonists.21 Accordingly, ADP failed to induce P-selectin exposure in any group.
As described above, RASGRP2 deletion impaired MK responses to 10 minutes of stimulation with both CRP and ADP, but not thrombin. To test for more subtle regulation by RASGRP2 as described for patient platelets, we stimulated MKs for a shorter time (2-3 minutes) before assessing activation. With this shorter time, deletion of RASGRP2 blunted integrin activation responses to thrombin to the same extent as CRP and convulxin (Figure 4A; supplemental Figure 6A shows the same data as MFI). P-selectin exposure in response to short-term treatment with GPVI agonists was also reduced by RASGRP2 KO, whereas P-selectin in response to thrombin was unaffected (Figure 4B; supplemental Figure 6B shows the same data as MFI). RAP1b total protein expression was increased in GP6 and RASGRP2 KO MKs (Figure 4C-D), but RAP1-GTP activation was abolished by GP6 KO after 1 minute of convulxin treatment and was impaired by RASGRP2 KO (Figure 4E-F), consistent with the described role of RASGRP2 as a guanine exchange factor (GEF).11
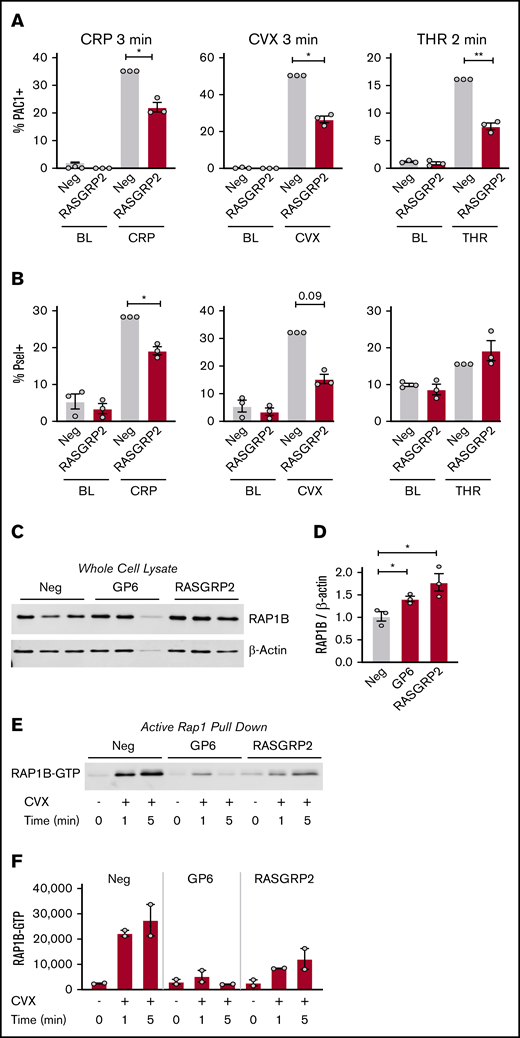
Integrin activation, P-selectin exposure, and RAP1 activation of RASGRP2 KO MKs in response to short treatment with platelet agonists. (A-B) Day 13 RASGRP2 CRISPR KO and control MKs were treated for 2 minutes with thrombin (THR, 1 U/mL) or 3 minutes with convulxin (CVX, 2 μg/mL) or CRP (2 μg/mL), stained for activated αIIb/β3 (PAC-1) and P-selectin, and analyzed by flow cytometry according to the gating strategy described in the legend for Figure 2B. Normalized mean ± SEM summaries of % PAC-1–positive MKs (A) or % P-selectin–positive MKs (B) (3 independent cords per group). (C-D) Western blot and densitometry analysis of total RAP1B protein in control and GP6 or RASGRP2 KO MKs (3 independent cords per group). One-way analysis of variance (ANOVA) with Dunnett’s adjustment for multiple comparisons. (E) RAP1-GTP was precipitated from equal numbers of negative control, GP6, or RASGRP2 KO MKs activated with convulxin, lysed at the time points indicated, and analyzed by western blot. Shown is a representative of 2 independent experiments from 2 different cords, which are summarized by the densitometry analysis in panel F. Paired Student t tests: *P < .05; **P < .01.
Integrin activation, P-selectin exposure, and RAP1 activation of RASGRP2 KO MKs in response to short treatment with platelet agonists. (A-B) Day 13 RASGRP2 CRISPR KO and control MKs were treated for 2 minutes with thrombin (THR, 1 U/mL) or 3 minutes with convulxin (CVX, 2 μg/mL) or CRP (2 μg/mL), stained for activated αIIb/β3 (PAC-1) and P-selectin, and analyzed by flow cytometry according to the gating strategy described in the legend for Figure 2B. Normalized mean ± SEM summaries of % PAC-1–positive MKs (A) or % P-selectin–positive MKs (B) (3 independent cords per group). (C-D) Western blot and densitometry analysis of total RAP1B protein in control and GP6 or RASGRP2 KO MKs (3 independent cords per group). One-way analysis of variance (ANOVA) with Dunnett’s adjustment for multiple comparisons. (E) RAP1-GTP was precipitated from equal numbers of negative control, GP6, or RASGRP2 KO MKs activated with convulxin, lysed at the time points indicated, and analyzed by western blot. Shown is a representative of 2 independent experiments from 2 different cords, which are summarized by the densitometry analysis in panel F. Paired Student t tests: *P < .05; **P < .01.
Because PAC-1 antibody staining and fibrinogen binding may not be redundant in megakaryocytes, ligand binding in response to convulxin stimulation was also assessed for each gene deletion using labeled fibrinogen as depicted by the representative flow plots in Figure 5A. Ten-minute stimulation with convulxin caused a significant increase in fibrinogen binding to MKs for negative control CRISPR-treated cells (Figure 5A-C). Consistent with the PAC-1 binding data, CRISPR deletion of ITGA2B, GP6, and RASGRP2 blunted MK binding to fibrinogen, whereas B2M deletion did not (Figure 5A-C). To assess spreading, the number of spread vs round or unspread MKs was assessed by microscopy (Figure 5D) after incubation on fibrinogen- or collagen-coated plates for 1 hour. Although deletion of RASGRP2 and B2M did not affect spreading on fibrinogen or collagen, deletion of ITGA2B significantly reduced the percentage of MKs that spread on fibrinogen and potentially (P = .10) on collagen (Figure 5E-F). Deletion of GP6 significantly reduced the percentage of MKs that spread on both fibrinogen and collagen (Figure 5E-F), which further confirms the dual receptor activities of GPVI previously shown using GPVI-deficient patient platelets.26
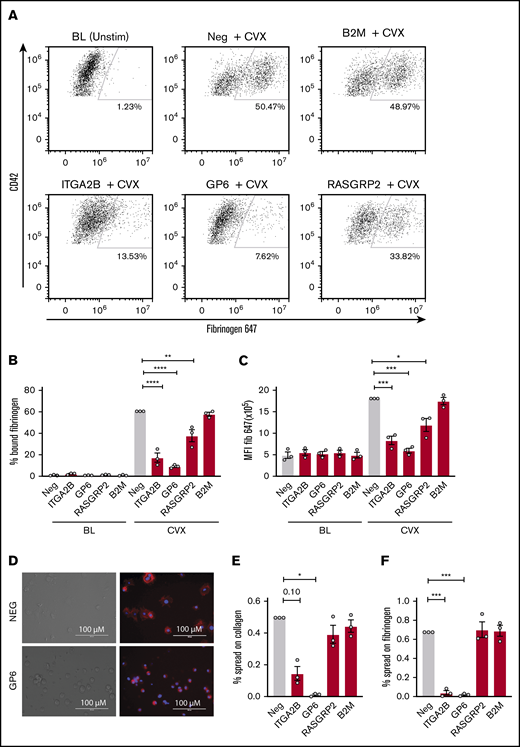
Ligand binding and spreading of CRISPR KO MKs in response to platelet agonists. Day 13 CRISPR KO and control MKs were treated for 10 minutes with convulxin (CVX, 2 μg/mL) in the presence of Alexa-Fluor 647–labeled fibrinogen and analyzed by flow cytometry. (A) Representative flow cytometry plots of unstimulated vs agonist-stimulated negative control or CRISPR KO MKs as indicated. Viable MKs were gated as described in the legend for Figure 2B and on MK marker CD42a-phycoerythrin (y-axis). The positive gate for labeled fibrinogen (x-axis) bound MKs was arbitrarily set with reference to unstimulated MKs. (B-C) Normalized mean ± SEM summaries of PAC-1–positive (B) or P-selectin–positive (C) cells (3 independent cords per group). Mixed effects analysis with Dunnett’s adjustment for multiple comparisons. (D) Representative image of spread and unspread control and GP6 KO MKs plated on collagen for 1 hour. (E-F) Mean ± SEM of the percentage of negative control or CRISPR KO MKs spread on collagen (E) or fibrinogen (F) for 1 hour. Shown are data from 3 cords with each sample plated in duplicate wells; >20 fields and >200 MKs were scored (blinded to treatment) per sample. Repeated measures ANOVA with Dunnett’s adjustment for multiple comparisons: *P < .05; **P < .01; ***P < .001; ****P < .0001.
Ligand binding and spreading of CRISPR KO MKs in response to platelet agonists. Day 13 CRISPR KO and control MKs were treated for 10 minutes with convulxin (CVX, 2 μg/mL) in the presence of Alexa-Fluor 647–labeled fibrinogen and analyzed by flow cytometry. (A) Representative flow cytometry plots of unstimulated vs agonist-stimulated negative control or CRISPR KO MKs as indicated. Viable MKs were gated as described in the legend for Figure 2B and on MK marker CD42a-phycoerythrin (y-axis). The positive gate for labeled fibrinogen (x-axis) bound MKs was arbitrarily set with reference to unstimulated MKs. (B-C) Normalized mean ± SEM summaries of PAC-1–positive (B) or P-selectin–positive (C) cells (3 independent cords per group). Mixed effects analysis with Dunnett’s adjustment for multiple comparisons. (D) Representative image of spread and unspread control and GP6 KO MKs plated on collagen for 1 hour. (E-F) Mean ± SEM of the percentage of negative control or CRISPR KO MKs spread on collagen (E) or fibrinogen (F) for 1 hour. Shown are data from 3 cords with each sample plated in duplicate wells; >20 fields and >200 MKs were scored (blinded to treatment) per sample. Repeated measures ANOVA with Dunnett’s adjustment for multiple comparisons: *P < .05; **P < .01; ***P < .001; ****P < .0001.
Novel platelet gene function analysis using CRISPR to delete COMMD7 in MKs
Several previous studies suggest an association between COMMD7 and platelet function (see Goodall et al,27 Galarneau et al,28 Salehe et al,29 and Guerrero30 and “Discussion”), but causative evidence is lacking. Therefore, COMMD7 was chosen to test CRIMSON as a tool for screening novel platelet gene functions. As shown in Figure 6A, COMMD7 transcript expression in MKs, as assessed by real-time PCR, is significantly reduced by CRISPR deletion. COMMD7 protein is expressed in platelets and MKs and deleted in MKs using CRISPR (Figure 6B). According to analysis with RNA sequencing of negative control and COMMD7 CRISPR MKs, transcripts of genes flanking COMMD7 were not reduced nor were any of the genes predicted as off-target binding sites (supplemental Table 1).
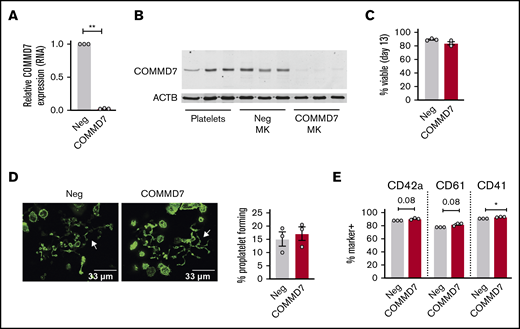
Effect of COMMD7 KO on megakaryopoiesis. (A) Real-time PCR analysis of COMMD7 RNA expression in COMMD7 CRISPR-treated day 13 MKs. Data are normalized to GAPDH (3 independent cords per group). (B) Western blot of COMMD7 protein in platelets from 3 healthy donors and in control and COMMD7 KO MKs from 3 independent cords per group. β-actin (ACTB) was used as loading control. (C) Percentage of viable negative control or COMMD7 KO cells on day 13 of culture. (D) Proplatelet formation on day 13 MKs plated overnight on fibrinogen and stained with Phalloidin 488. Shown are representative images of 10 random images taken from each of 3 cords. The percentage of control or COMMD7 KO MKs making proplatelets on day 13 was counted blinded to group and summarized in the bar graph on the right. (E) Flow cytometry analysis of MK maturation markers on day 13 control or COMMD7 MKs. Viable MKs were gated as in Figure 2B. Shown is the mean ± SEM of the percent of cells (y-axis) expressing MK maturation markers. Student t test: *P < .05; **P < .01.
Effect of COMMD7 KO on megakaryopoiesis. (A) Real-time PCR analysis of COMMD7 RNA expression in COMMD7 CRISPR-treated day 13 MKs. Data are normalized to GAPDH (3 independent cords per group). (B) Western blot of COMMD7 protein in platelets from 3 healthy donors and in control and COMMD7 KO MKs from 3 independent cords per group. β-actin (ACTB) was used as loading control. (C) Percentage of viable negative control or COMMD7 KO cells on day 13 of culture. (D) Proplatelet formation on day 13 MKs plated overnight on fibrinogen and stained with Phalloidin 488. Shown are representative images of 10 random images taken from each of 3 cords. The percentage of control or COMMD7 KO MKs making proplatelets on day 13 was counted blinded to group and summarized in the bar graph on the right. (E) Flow cytometry analysis of MK maturation markers on day 13 control or COMMD7 MKs. Viable MKs were gated as in Figure 2B. Shown is the mean ± SEM of the percent of cells (y-axis) expressing MK maturation markers. Student t test: *P < .05; **P < .01.
On day 13 of culture, COMMD7 KO MKs were similar to controls in viability (Figure 6C), were large and polyploid (supplemental Figure 7A), contained granules, had an extensive demarcation membrane system (supplemental Figure 7B), made proplatelets (Figure 6D), and expressed normal levels of maturation markers (Figure 6E). Deletion of COMMD7 significantly reduced integrin activation in response to both convulxin and thrombin (Figure 7A; supplemental Figure 8A-B). Fibrinogen binding in response to convulxin (Figure 7B; supplemental Figure 8C) was also impaired, whereas spreading on fibrinogen or collagen was unaffected (Figure 7C). A more pronounced impairment was observed for P-selectin translocation (Figure 7D-E; supplemental Figure 8D-E). This more pronounced phenotype could be partly explained by a modest reduction in total P-selectin in COMMD7 KO MKs compared with controls (supplemental Figure 8F). However, targeted experiments testing CD63 exposure, which marks dense granules and lysosomes, suggested additional granule effects beyond P-selectin expression (Figure 7F; supplemental Figure 8G). COMMD7 effects appeared downstream or independently of PKC activation because COMMD7 deletion did not reduce phosphorylation of PKC substrates in response to convulxin or thrombin (supplemental Figure 9). Together, data from the CRIMSON analysis indicate that COMMD7 causally affects platelet-like functional responses in MKs through multiple pathways.
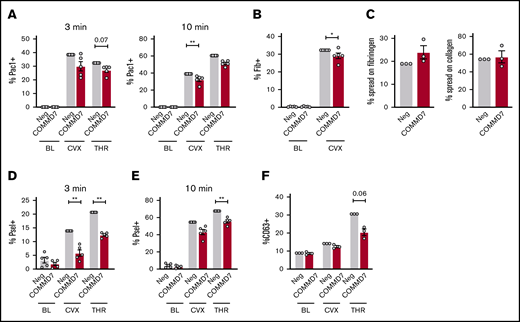
Effect of COMMD7 KO on platelet functional responses in MKs. (A-B,D-F) Negative control or COMMD7 KO day 13 MKs were kept at baseline or stimulated with convulxin (2 μg/mL) or thrombin (1 U/mL) for 3 or 10 minutes as indicated and analyzed by flow cytometry. Viable MKs were gated as in Figure 2B. Shown is the normalized mean ± SEM of percent positive cells for PAC-1 (A), labeled fibrinogen (B), surface P-selectin (D-E) , or surface CD63 (F) (3-5 independent cords per group). (C) Mean ± SEM of the percentage of control or CRISPR KO MKs spread after 1 hour on collagen or fibrinogen. Shown are samples from 3 cords with each sample plated in duplicate wells; >20 fields and >200 MKs were scored (blinded to treatment) per sample. Paired Student t tests: *P < .05; **P < .01.
Effect of COMMD7 KO on platelet functional responses in MKs. (A-B,D-F) Negative control or COMMD7 KO day 13 MKs were kept at baseline or stimulated with convulxin (2 μg/mL) or thrombin (1 U/mL) for 3 or 10 minutes as indicated and analyzed by flow cytometry. Viable MKs were gated as in Figure 2B. Shown is the normalized mean ± SEM of percent positive cells for PAC-1 (A), labeled fibrinogen (B), surface P-selectin (D-E) , or surface CD63 (F) (3-5 independent cords per group). (C) Mean ± SEM of the percentage of control or CRISPR KO MKs spread after 1 hour on collagen or fibrinogen. Shown are samples from 3 cords with each sample plated in duplicate wells; >20 fields and >200 MKs were scored (blinded to treatment) per sample. Paired Student t tests: *P < .05; **P < .01.
Discussion
Our CRIMSON approach enables simple, rapid, reproducible, and efficient gene deletions in primary human MKs followed by functional testing without selection or sorting. The ability to efficiently delete and evaluate platelet function genes in primary MKs fills a critical gap in the field of study regarding platelets. Genetic association studies have implicated numerous genes in platelet function, but the data are usually insufficient to establish causation. Because of this, gene studies of platelet disease rely on curated lists of genes and variants classified according to the layers of evidence for causation. One such curation includes 61 Tier 1 genes related to platelet function or formation recently published by the Scientific and Standardization Committee (SSC) of the International Society on Thrombosis and Haemostasis (ISTH).2 Molecular functional studies and mouse models are considered an important layer of evidence for this classification process, especially when genetic evidence is limited to a few families. Unfortunately, studies of platelet gene function have lagged far behind genetic discovery. Many Tier 2 and Tier 3 genes and numerous others from association studies await functional validation.
By searching the literature, we identified 1 such gene, COMMD7, as a gene that is associated with platelet function and disease phenotypes, but there was no additional evidence for causation. COMMD7 RNA expression in platelets is abundant (top 10% of platelet transcripts) and consistent over time (repeatability index, 0.79),31 and it was positively associated with in vitro platelet functional responses to collagen receptor agonists as measured by P-selectin exposure and fibrinogen binding.27 Single nucleotide polymorphisms near COMMD7 were associated with cardiovascular disease27,29 and potentially platelet aggregation in children.30 A significant association was identified for a single nucleotide polymorphism near COMMD7 and acute chest syndrome in sickle cell disease.28 Whole-body morpholino silencing of COMMD7 in zebrafish resulted in reduced thrombus formation.27 Despite these associations, very little is known about the cellular functions of COMMD7. We used CRIMSON to confirm a role for COMMD7 in select platelet functions in MKs, including integrin activation and degranulation, with a more pronounced effect on P-selectin exposure. Future in vitro and in vivo studies are needed to evaluate additional contributions and mechanisms of COMMD7 in MKs and platelets.
It is well known that platelet responses vary substantially between individuals, with a significant contribution by genetics. One benefit of using primary cells is the ability to test the effect of gene deletions in the context of different genetic backgrounds on individual cord blood units, which is not as straightforward for cell lines. The use of bulk cultures of CRISPR KO primary cells also avoids problems of genetic drift that can accompany iPSCs, cell lines, and especially cultures grown from single clones. As a trade-off, KOs in bulk cultures may suffer from purity, especially for transcripts and proteins that may be very stable. We expect this will be problematic for only a small subset of genes, given the exceptional level of knock-down of RNA and protein levels achieved for all genes tested.
Side-by-side simultaneous evaluations of multiple different KOs can be also accomplished in primary cells of the same genetic background (ie, from the same unit of cord blood) if desired. For example, from 1 unit of cord blood (typically ∼50 mL) we assessed the effect of 5 different gene KOs, yielding 2.8 ± 0.6 × 106 (mean ± standard deviation) MKs per KO on day 13 for functional assays. Side-by-side evaluations are important for assessing the relative contributions of genes to specific pathways. Whereas GP6 and ITGA2B KO resulted in pronounced, nearly dichotomous (all or none) MK activation phenotypes, RASGRP2 KO activation phenotypes were more subtle, as might be expected from published platelet studies.11,24 From a methodology standpoint, this indicates that nuanced or quantitative phenotypes might be identified using CRIMSON with careful consideration of the appropriate read-out, agonist choice, dose, or time point, all of which are important considerations for evaluating new gene functions in MKs and platelets.
In many of our experiments, we deleted B2M as a control for CRISPR deletion. Beyond its usefulness as a control, deletion of B2M in platelets has long been explored as a method of circumventing the problem of allogeneic platelet transfusions in refractory individuals.32 Antibiotic selection or sorting of lentivirus delivered RNA interference, transfected transcription activator-like effector nucleases (TALENs), most recently CRISPR/Cas9 has been used to obtain B2M deletion in CD34+ cells or iPSCs for differentiation into HLA class 1–deficient MKs and production of platelet-like particles.32-35 Platelet B2M has also recently been implicated in immune regulation.36,37
We suggest the use of CRIMSON as a screening tool to complement mouse models and other in vitro and in vivo platelet studies. As with any model, there are limitations. Obvious and inevitable differences between in vivo circulating adult platelets and in vitro MKs from cord blood should be carefully considered when interpreting CRIMSON results. For example, in our studies, we noted an unexplained blunted response to ADP for MKs compared with platelets. Likewise, other platelet gene functions sensitive to development, cell size, platelet age, shear, or presence of plasma components might yield different results in an MK KO model vs a platelet. Genes that affect individual proteins and RNA can also differ in expression between MKs and platelets. Quantitative proteome and transcriptome comparisons between platelets and MKs may be helpful in this regard. Notwithstanding model limitations, our findings support the concept that primary human CRISPR-modified MKs can be used in certain conditions as surrogates to study platelet genes. In addition to the functional read-outs tested in this study, other aspects of platelet function such as leukocyte binding, clot retraction, aggregate formation, or adhesion and thrombus formation under flow may also be plausibly tested with MKs, but this remains to be determined. Alternatively, platelet-like particles could be harvested from CRISPR-modified MKs for further testing in vitro or in vivo.33,35,38,39
Indirect and off-target effects are also possible with CRISPR. For example, MK maturity and viability may indirectly affect functional responses and should be noted for each new gene evaluated. CRISPR might also cut in unintended places. To address this for COMMD7, we used RNA sequencing to analyze surrounding genes and genes harboring predicted off-target sequences. Residual off-target concerns could also be alleviated by using multiple CRISPRs targeting different sequences in the same gene. Rescue experiments might also be performed.
In summary, as demonstrated for ITGA2B, GP6, RASGRP2, B2M, and COMMD7, we expect that the CRIMSON approach developed in this study will accelerate the functional assessment of additional platelet genes and facilitate mechanistic evaluation of gene phenotype associations.
To request data, please e-mail Jesse W. Rowley at jesse.rowley@u2m2.utah.edu.
Acknowledgments
The authors thank Diana Lim for preparing the figures, critical comments, and consultation regarding effective display of the images and Robert Campbell and Frederik Denorme for reviewing the manuscript.
This work was supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (HL144957 [J.W.R.] and HL142804 [M.T.R.]), by grants from the American Heart Association (18POST34030020 [B.K.M.] and 20POST35210319 [E.M.]), by a Merit Review Award (I01 CX001696) (M.T.R.) from the US Department of Veterans Affairs Clinical Sciences R&D Service, and by resources and the use of facilities at the George E. Wahlen VA Medical Center, Salt Lake City, UT.
The contents of this article do not represent the views of the US Department of Veterans Affairs or the United States Government.
Authorship
Contribution: E.M. and S.B. designed and performed the research and analyzed the data; S.J. performed research and analyzed the data; Y.K., B.K.M., O.L., I.P.-I., E.T., and N.D.T. performed the research; M.T.R. and P.F.B. designed the research; and J.W.R. designed and performed the research, analyzed the data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jesse W. Rowley, Eccles Institute of Human Genetics, University of Utah Health Sciences Center, 15 North 2030 East, Room 4220, Salt Lake City, UT 84112; e-mail: jesse.rowley@u2m2.utah.edu.

