

Key Points
Defibrotide is associated with encouraging responses in the real world, including VOD/SOS after MAC as well as RIC allogeneic HSCT.
Early diagnosis, prompt initiation of defibrotide, and minimization of dosing interruptions may be key to successful treatment of VOD/SOS.

Abstract
Hepatic veno-occlusive disease or sinusoidal obstructive syndrome (VOD/SOS) is a life-threatening complication of hematopoietic stem cell transplantation (HSCT). Defibrotide is the only medication approved by the US Food and Drug Administration for the management of severe VOD/SOS after HSCT. We report our center’s experience with commercially available defibrotide as treatment of patients with VOD/SOS. We retrospectively identified 28 cases of VOD/SOS, based on the European Society for Blood and Marrow Transplantation criteria, from March 2016 through June 2019. The median day of VOD/SOS onset was 25 days (range, 8-69 days), and defibrotide was initiated on day of diagnosis in 71% of patients. Complete resolution of VOD/SOS occurred in 75% of patients. Day 100 survival was 64% for all HSCT patients and 53% for those with very severe VOD/SOS. Response rates and survival were similar in patients with VOD/SOS after myeloablative or reduced-intensity chemotherapy HSCT. Therapy-related adverse events were mild and included hematuria (43%), epistaxis (18%), and hypotension (11%). Severe hemorrhagic adverse events occurred in 2 patients (pulmonary hemorrhage and upper gastrointestinal hemorrhage; 7%) and both in the setting of progressive VOD/SOS. Early diagnosis, prompt initiation of defibrotide, and minimization of dosing interruptions may be key to successful treatment of VOD/SOS.
Introduction
Hepatic veno-occlusive disease/sinusoidal obstructive syndrome (VOD/SOS) is a potentially life-threatening complication following hematopoietic stem cell transplantation (HSCT). In a retrospective analysis of 135 pooled studies, there was a 13.7% overall incidence of VOD/SOS, although incidence rates after myeloablative conditioning (MAC) HSCT have declined over the last decade.1 With the use of reduced-intensity chemotherapy (RIC) regimens, the rate is even lower.2-4 Estimated day 100 mortality rates have been reported as 9%, 23%, and 98% in patients categorized as having mild, moderate, or severe VOD/SOS.5 Diagnosis of VOD/SOS is traditionally made by using Baltimore and/or modified Seattle criteria, with both sets of criteria based on the clinical triad of weight gain/ascites, hepatomegaly/right upper quadrant pain, and hyperbilirubinemia. These “classical” criteria were developed decades ago from cohorts of adult patients who developed early hepatic VOD/SOS (within day 21 of HSCT) after MAC regimens such as busulfan/cyclophosphamide, cyclophosphamide/total body irradiation, and cyclophosphamide/carmustine/etoposide and within a defined period post-HSCT (≤21 days). Their applicability in the diagnosis of VOD/SOS in the pediatric setting or modern era of RIC and reduced-toxicity ablative conditioning regimens is less clear. Recently, the European Society for Blood and Marrow Transplantation (EBMT) has proposed newer sets of VOD/SOS diagnostic criteria that added such criteria for pediatrics, as well as adults who develop VOD/SOS beyond day 21 and those who do not meet classical criteria but have VOD/SOS based on ultrasound evidence.6 In addition, patients with VOD/SOS may not present with hyperbilirubinemia (total bilirubin level >2 mg/dL) but have other features consistent with VOD/SOS defined as anicteric VOD/SOS.7
Defibrotide was approved by the US Food and Drug Administration (FDA) in March 2016 for the treatment of VOD/SOS in HSCT patients with renal or pulmonary dysfunction. It is a mixture of single- and double-stranded oligonucleotides derived from porcine intestinal mucosal DNA.9 Although its mechanism of action is not fully elucidated, in vitro, it seems to augment plasmin enzymatic activity to hydrolyze fibrin clots, reduces endothelial cell activation, increases fibrinolysis through increased tissue plasminogen activator and thrombomodulin expression, and decreases von Willebrand factor and plasminogen activator inhibitor-1 expression. The dose-finding phase 2 trial recommended an optimal dosage of 25 mg/kg per day in 4 divided doses for at least 21 days.10,11
The defibrotide registration trial, a phase 3, multicenter, open-label study, assessed the efficacy and safety of defibrotide 25 mg/kg per day at day 100 post-HSCT in patients with hepatic VOD/SOS with advanced multiorgan dysfunction (MOD). It used a novel contemporaneous historical control methodology, incorporating propensity analysis to generate a rigorous comparator. The day 100 observed survival rate was 38.2% vs 25% in the historical matched control arm (P = .0109). The toxicity profile of defibrotide compared with that of the historical control showed that pulmonary alveolar and gastrointestinal hemorrhages occurred in 11.8% vs 15.6% and 7.8% vs 9.4%, respectively. A large, expanded access program under a treatment investigational new drug application in >1000 patients confirmed this favorable safety profile and also reported efficacy across various settings, including severe disease and advanced multiorgan failure.4,12
Since the FDA approval of defibrotide, however, there have been no published data regarding the “real-world” efficacy and safety of the drug, particularly in the context of the newly established EBMT criteria. Here we report our center’s experience with commercially available defibrotide for the treatment of patients with VOD/SOS from March 2016 through June 2019.
Methods
Study design and participants
This study was conducted at the Dana-Farber Cancer Institute/Brigham and Women’s Hospital and was approved by the Dana-Farber/Harvard Cancer Center Institutional Review Board. The pharmacy records were screened for all patients aged ≥18 years who received at least 1 dose of commercial defibrotide for the management of hepatic VOD/SOS between March 2016 and June 2019. Patients were excluded if they had received prophylactic defibrotide without developing VOD/SOS or were administered defibrotide for the treatment of drug-induced VOD/SOS without HSCT. After the diagnosis of VOD/SOS, defibrotide was initiated at the discretion of the transplant physician and administered at 25 mg/kg per day in 4 divided doses. Patients’ baseline weight before transplant was used to calculate the final dose of defibrotide.
The primary objective of the current study was to assess the efficacy of defibrotide as represented by survival at day 100 and resolution of VOD/SOS. Our secondary objective was to describe and assess treatment-related adverse events.
Response and outcome definitions
VOD/SOS diagnosis and severity assessment were defined according to the EBMT consensus guidelines.6 Treating physicians also relied on radiologic ultrasound criteria before day 21 to aid in the early diagnosis of VOD/SOS. Ultrasound evidence for VOD/SOS included slow, bidirectional, or reversal of flow in the portal vein supporting the presence of portal hypertension. Survival at day 100 and VOD/SOS symptom resolution were used for the analysis. Weekly ultrasounds with Doppler studies were performed to assess for improvement and normalization of flow for those who had abnormal flow at diagnosis. Renal function and respiratory and volume status were also assessed routinely for normalization by the end of therapy. Resolution of VOD/SOS was defined as: total bilirubin level <2 mg/dL; serum creatinine level <1.5 times baseline level; creatinine clearance >80% of initial rate and dialysis independent; and oxygen saturation >90% on room air, no supplemental oxygen required, or ventilator independence.
Statistical methods
Data analysis was primarily descriptive using Fisher’s exact test for group comparison as appropriate. Response to defibrotide treatment, overall survival (OS) and progression-free survival (PFS), were the outcomes of interest. OS was defined as time from the date of VOD diagnosis to death. PFS was defined as time from the date of VOD diagnosis to relapse or death, whichever occurred first. The Kaplan-Meier method was used to estimate OS and PFS, and the log-rank test was used to compare OS and PFS for baseline characteristics. Univariable Cox regression analysis was used to identify risk factors for OS and PFS. Response was treated as a time-dependent variable in the Cox model, and Makuch-Simon curves were generated to depict the time dependency graphically.13,14
All P values were two sided, and the significance level was set to .05. Multiplicity was not considered. All analyses were performed by using SAS 9.3 (SAS Institute Inc., Cary, NC) and R version 3.2.2 (the CRAN project, www.cran.r-project.org).
Results
Patient characteristics and study cohort
We identified 38 patients who received at least 1 dose of commercial defibrotide from March 2016 through June 2019. Ten patients were excluded for the following reasons: 4 for receiving prophylactic defibrotide, 4 for drug-induced VOD/SOS without HSCT (inotuzumab ozogamicin [n = 1], gemtuzumab ozogamicin [n = 2], and nivolumab [n = 1]), 1 for receiving prophylactic defibrotide at a dose of 20 mg/kg per day, and 1 for decreased portal vein flow not associated with HSCT. Twenty-eight patients met the inclusion criteria and were included in the analysis. Two patients, despite receiving prophylactic defibrotide, developed VOD/SOS and were included in the analysis. During the study period, 909 allogeneic and 726 autologous HSCTs were performed at our center. Overall incidence of VOD/SOS at our center was 1.7%. Incidence of VOD/SOS among patients who received an allogeneic HSCT was 3.1% (MAC, 3.6%; RIC, 2.7%). All patients who developed VOD/SOS received an allogeneic HSCT, with 12 patients receiving a MAC regimen and 16 patients receiving an RIC regimen. The most common graft-versus-host disease (GVHD) prophylaxis regimen was tacrolimus and sirolimus (32%) and tacrolimus, sirolimus, and methotrexate (29%). Acute lymphoblastic leukemia (29%) and acute myeloid leukemia (25%) were the most common diagnoses. Four of the 28 patients had previous exposure to inotuzumab ozogamicin. The median age was 58 years (range, 26-75 years). Table 1 summarizes patients’ baseline characteristics.
Baseline characteristics
| Characteristic | All HSCT (N = 28) | Mild-moderate-severe VOD/SOS (n = 11) | Very severe VOD/SOS (n = 17) |
|---|---|---|---|
| Sex, n (%) | |||
| Male | 22 (79) | 9 (82) | 13 (76) |
| Female | 6 (21) | 2 (18) | 4 (24) |
| Age at time of HSCT, mean (range), y | 58 (26-75) | 55 (29-72) | 55 (26-75) |
| Race, n (%) | |||
| White | 22 (79) | 11 (100) | 11 (65) |
| Asian | 2 (7) | 0 | 2 (12) |
| Unknown | 4 (14) | 0 | 4 (23) |
| Body mass index, mean (range), kg/m2 | 25.35 (17.5-35.5) | 25.78 (20.55-31.4) | 25.1 (17.6-35.5) |
| Transplant type, n (%) | |||
| Allogeneic | 28 (100) | 11 (100) | 17 (100) |
| Donor characteristics, n (%) | |||
| Matched, related | 5 (18) | 2 (18) | 3 (18) |
| Matched, unrelated | 17 (62) | 7 (64) | 10 (58) |
| Mismatched, related | 3 (10) | 0 | 3 (18) |
| Mismatched, unrelated | 3 (10) | 2 (18) | 1 (6) |
| Conditioning agents, n (%) | |||
| Cyclophosphamide/TBI 1200 cGy | 2 (7) | 1 (9) | 1 (6) |
| Cyclophosphamide, fludarabine/TBI 200 cGy | 1 (4) | 1 (9) | 0 |
| Myeloablative busulfan and fludarabine | 10 (36) | 1 (9) | 9 (53) |
| Reduced-intensity busulfan and fludarabine | 12 (43) | 7 (64) | 5 (29) |
| Melphalan and fludarabine | 3 (10) | 1 (9) | 2 (12) |
| GVHD prophylaxis, n (%) | |||
| Cyclophosphamide | 2 (7) | 1 (9) | 1 (6) |
| Cyclophosphamide, tacrolimus, and mycophenolate mofetil | 3 (10) | 1 (9) | 2 (12) |
| Tacrolimus and sirolimus | 9 (32) | 2 (18) | 7 (40) |
| Tacrolimus and methotrexate | 5 (18) | 2 (18) | 3 (18) |
| Tacrolimus, sirolimus, and methotrexate | 8 (29) | 5 (46) | 3 (18) |
| Tacrolimus and mycophenolate mofetil | 1 (4) | 0 | 1 (6) |
| Primary disease, n (%) | |||
| AML | 7 (25) | 2 (18) | 6 (34) |
| ALL | 8 (29) | 4 (36) | 3 (18) |
| MDS | 6 (22) | 3 (27) | 3 (18) |
| MPN | 3 (10) | 1 (9) | 2 (12) |
| Mixed MDS/MPN | 1 (4) | 0 | 1 (6) |
| NHL | 3 (10) | 1 (9) | 2 (12) |
| Type of conditioning regimen, n (%) | |||
| MAC | 12 (43) | 2 (18) | 10 (59) |
| RIC | 16 (57) | 9 (82) | 7 (41) |
| Prior HSCTs, n (%) | |||
| None | 24 (86) | 8 (73) | 16 (94) |
| ≥1 | 4 (14) | 3 (27) | 1 (6) |
| VOD/SOS onset after day 21, n (%) | 16 (57) | 7 (64) | 9 (53) |
| Time from diagnosis to start of defibrotide, n (%) | |||
| Day of diagnosis | 20 (71) | 8 (73) | 12 (70) |
| Day 1 | 4 (14) | 1 (9) | 3 (18) |
| Day 2 | 1 (4) | 1 (9) | 0 |
| Day 3 | 1 (4) | 0 | 1 (6) |
| Prophylactic defibrotide with VOD | 2 (7) | 1 (9) | 1 (6) |
| Immunoconjugate treatment history, n (%) | |||
| Inotuzumab ozogamicin | 4 (14) | 3 (27) | 1 (6) |
| Gemtuzumab ozogamicin | 0 | 0 | 0 |
| History of liver impairment, n (%) | |||
| Hepatitis C infection | 1 (4) | 0 | 1 (6) |
| Alcohol dependence | 2 (7) | 1 (9) | 1 (6) |
| Oncologic liver disease involvement | 1 (4) | 0 | 1 (6) |
| Gilbert’s syndrome | 1 (4) | 1 (9) | 0 |
| Fatty liver (nonalcoholic) | 1 (4) | 1 (9) | 0 |
| Cholangitis | 1 (4) | 0 | 1 (6) |
| Inotuzumab-induced transaminitis | 1 (4) | 1 (9) | 0 |
| Graft source | |||
| Bone marrow | 6 (21) | 1 (9) | 5 (29) |
| Peripheral blood | 22 (79) | 10 (91) | 12 (71) |
| Characteristic | All HSCT (N = 28) | Mild-moderate-severe VOD/SOS (n = 11) | Very severe VOD/SOS (n = 17) |
|---|---|---|---|
| Sex, n (%) | |||
| Male | 22 (79) | 9 (82) | 13 (76) |
| Female | 6 (21) | 2 (18) | 4 (24) |
| Age at time of HSCT, mean (range), y | 58 (26-75) | 55 (29-72) | 55 (26-75) |
| Race, n (%) | |||
| White | 22 (79) | 11 (100) | 11 (65) |
| Asian | 2 (7) | 0 | 2 (12) |
| Unknown | 4 (14) | 0 | 4 (23) |
| Body mass index, mean (range), kg/m2 | 25.35 (17.5-35.5) | 25.78 (20.55-31.4) | 25.1 (17.6-35.5) |
| Transplant type, n (%) | |||
| Allogeneic | 28 (100) | 11 (100) | 17 (100) |
| Donor characteristics, n (%) | |||
| Matched, related | 5 (18) | 2 (18) | 3 (18) |
| Matched, unrelated | 17 (62) | 7 (64) | 10 (58) |
| Mismatched, related | 3 (10) | 0 | 3 (18) |
| Mismatched, unrelated | 3 (10) | 2 (18) | 1 (6) |
| Conditioning agents, n (%) | |||
| Cyclophosphamide/TBI 1200 cGy | 2 (7) | 1 (9) | 1 (6) |
| Cyclophosphamide, fludarabine/TBI 200 cGy | 1 (4) | 1 (9) | 0 |
| Myeloablative busulfan and fludarabine | 10 (36) | 1 (9) | 9 (53) |
| Reduced-intensity busulfan and fludarabine | 12 (43) | 7 (64) | 5 (29) |
| Melphalan and fludarabine | 3 (10) | 1 (9) | 2 (12) |
| GVHD prophylaxis, n (%) | |||
| Cyclophosphamide | 2 (7) | 1 (9) | 1 (6) |
| Cyclophosphamide, tacrolimus, and mycophenolate mofetil | 3 (10) | 1 (9) | 2 (12) |
| Tacrolimus and sirolimus | 9 (32) | 2 (18) | 7 (40) |
| Tacrolimus and methotrexate | 5 (18) | 2 (18) | 3 (18) |
| Tacrolimus, sirolimus, and methotrexate | 8 (29) | 5 (46) | 3 (18) |
| Tacrolimus and mycophenolate mofetil | 1 (4) | 0 | 1 (6) |
| Primary disease, n (%) | |||
| AML | 7 (25) | 2 (18) | 6 (34) |
| ALL | 8 (29) | 4 (36) | 3 (18) |
| MDS | 6 (22) | 3 (27) | 3 (18) |
| MPN | 3 (10) | 1 (9) | 2 (12) |
| Mixed MDS/MPN | 1 (4) | 0 | 1 (6) |
| NHL | 3 (10) | 1 (9) | 2 (12) |
| Type of conditioning regimen, n (%) | |||
| MAC | 12 (43) | 2 (18) | 10 (59) |
| RIC | 16 (57) | 9 (82) | 7 (41) |
| Prior HSCTs, n (%) | |||
| None | 24 (86) | 8 (73) | 16 (94) |
| ≥1 | 4 (14) | 3 (27) | 1 (6) |
| VOD/SOS onset after day 21, n (%) | 16 (57) | 7 (64) | 9 (53) |
| Time from diagnosis to start of defibrotide, n (%) | |||
| Day of diagnosis | 20 (71) | 8 (73) | 12 (70) |
| Day 1 | 4 (14) | 1 (9) | 3 (18) |
| Day 2 | 1 (4) | 1 (9) | 0 |
| Day 3 | 1 (4) | 0 | 1 (6) |
| Prophylactic defibrotide with VOD | 2 (7) | 1 (9) | 1 (6) |
| Immunoconjugate treatment history, n (%) | |||
| Inotuzumab ozogamicin | 4 (14) | 3 (27) | 1 (6) |
| Gemtuzumab ozogamicin | 0 | 0 | 0 |
| History of liver impairment, n (%) | |||
| Hepatitis C infection | 1 (4) | 0 | 1 (6) |
| Alcohol dependence | 2 (7) | 1 (9) | 1 (6) |
| Oncologic liver disease involvement | 1 (4) | 0 | 1 (6) |
| Gilbert’s syndrome | 1 (4) | 1 (9) | 0 |
| Fatty liver (nonalcoholic) | 1 (4) | 1 (9) | 0 |
| Cholangitis | 1 (4) | 0 | 1 (6) |
| Inotuzumab-induced transaminitis | 1 (4) | 1 (9) | 0 |
| Graft source | |||
| Bone marrow | 6 (21) | 1 (9) | 5 (29) |
| Peripheral blood | 22 (79) | 10 (91) | 12 (71) |
ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; MDS, myelodysplastic syndrome; MPN, myeloproliferative neoplasms; NHL, non-Hodgkin lymphoma; TBI, total body irradiation.
VOD/SOS diagnosis
All patients met EBMT diagnosis criteria, 4 patients met strict Baltimore criteria (before day 21) at diagnosis, and 4 patients technically also met Baltimore criteria but were beyond day 21.6 Table 2 summarizes patients’ baseline clinical symptoms at diagnosis. All patients undergoing allogeneic HSCT at our center received ursodeoxycholic acid as VOD/SOS prophylaxis starting 1 week before conditioning and continued until at least day 60 to 100.
Clinical symptoms at time of VOD/SOS diagnosis
| Symptom | All HSCT (N = 28) | Mild-moderate-severe VOD/SOS (n = 11) | Very severe VOD/SOS (n = 17) |
|---|---|---|---|
| Ascites, n (%) | 26 (93) | 10 (91) | 16 (94) |
| Painful hepatomegaly, n (%) | 17 (61) | 7 (64) | 10 (59) |
| Renal dysfunction, n (%) | 21 (75) | 6 (55) | 15 (88) |
| Reversal of flow, n (%) | 16 (57) | 5 (45) | 11 (65) |
| Pulmonary dysfunction, n (%) | 6 (21) | 0 | 6 (35) |
| Symptom | All HSCT (N = 28) | Mild-moderate-severe VOD/SOS (n = 11) | Very severe VOD/SOS (n = 17) |
|---|---|---|---|
| Ascites, n (%) | 26 (93) | 10 (91) | 16 (94) |
| Painful hepatomegaly, n (%) | 17 (61) | 7 (64) | 10 (59) |
| Renal dysfunction, n (%) | 21 (75) | 6 (55) | 15 (88) |
| Reversal of flow, n (%) | 16 (57) | 5 (45) | 11 (65) |
| Pulmonary dysfunction, n (%) | 6 (21) | 0 | 6 (35) |
We have adopted the use of ultrasound evidence at our center as part of the EBMT diagnostic criteria even in patients before day 21. All patients had a liver ultrasound with Doppler studies at diagnosis. The most common findings supporting the VOD/SOS diagnosis included ascites (93%), portal venous flow abnormalities (57%), and hepatomegaly (61%). Abnormal portal venous flow was detected by Doppler ultrasound in 16 patients: 10 reversed flow, 5 slow flow (defined as peak velocity ≤15 cm/s), and 1 bidirectional flow.
Liver biopsy samples were obtained in 5 patients. They were all diagnosed beyond day 21 (median, 30 days; range, 22-69 days), had ascites, and experienced renal dysfunction but did not display flow reversal on ultrasound.
Transthoracic echocardiogram was obtained in 10 patients to rule out a competing diagnosis of heart failure, and 9 patients had a normal ejection fraction and ventricular size. None had right ventricular dysfunction or pulmonary hypertension.
VOD/SOS was diagnosed at a median of 25 days (range, 8-69 days) after HSCT. Sixteen patients (57%) developed VOD/SOS beyond day 21; of these patients, 69% received an RIC regimen and 75% received sirolimus as part of their GVHD regimen. This contrasts with those who developed VOD/SOS before day 21, with 58% receiving a MAC regimen and only 42% receiving sirolimus as part of their GVHD regimen.
Among the 28 patients in this study, 15 had anicteric VOD/SOS. Of these, 9 (60%) received an RIC transplant. All the patients with anicteric VOD/SOS had ascites, 12 (80%) had ultrasound evidence of VOD/SOS, and 11 (73%) had renal dysfunction. Three of these 15 anicteric VOD/SOS cases were diagnosed by using liver biopsy results.
VOD/SOS disease severity
Of the 4 patients who strictly met the Baltimore criteria before day 21, one had severe VOD/SOS and 3 had very severe disease according to EBMT severity criteria. In these 4 patients, defibrotide therapy was started on the day of VOD/SOS diagnosis. Among the remaining patients who met EBMT criteria for diagnosis only, 1, 6, and 4 had mild, moderate, and severe disease, respectively.
Defibrotide treatment and response
Defibrotide was administered for a median duration of 19 days (range, 2-45 days), and therapy was initiated on day of diagnosis in 71% of patients, 14% on day 2, and 4% each on days 3 and 4. Two patients were on prophylactic defibrotide during conditioning before transplant, and defibrotide was continued when these patients subsequently developed VOD/SOS.
Complete resolution of VOD/SOS was observed in 21 patients (75%), with a median time to resolution of 14 days (range, 2-106 days). Response to defibrotide as a time-dependent variable in the Cox model was significantly associated with OS (hazard ratio, 0.14; 95% confidence interval, 0.03-0.69; P = .016) and PFS (hazard ratio, 0.16; 95% confidence interval, 0.04-0.84; P = .029). Sixteen patients had abnormal portal vein flow on ultrasound at diagnosis. Of these, 14 patients had clinical resolution of VOD/SOS and normalization of flow on ultrasound that occurred at a median of 14 days from defibrotide initiation. Four patients who met strict Baltimore criteria for VOD/SOS diagnosis before day 21 died of multiorgan failure and VOD/SOS. In these 4 patients, the mean total bilirubin and percent weight gain at diagnosis were 3.48 mg/dL and 8.7%, respectively.
Survival after defibrotide treatment
For the entire cohort, the median follow-up time from VOD/SOS onset was 18 months (range, 11-33 months), and 18 patients (64%) were alive at day 100 after VOD/SOS diagnosis. One-year treatment-related mortality was 39%. Patients who developed mild, moderate, and severe VOD/SOS, based on the EBMT criteria, had improved day 100 survival compared with those who exhibited very severe symptoms (82% vs 53%; P = .23), although the difference was not statistically significant.6 Seven patients had MOD, defined as involving renal and/or pulmonary dysfunction. Of those with MOD (n = 7), 4 (57%) patients were alive at day 100. Day 100 survival was 58% and 75% among patients who received MAC and RIC, respectively, and there was no difference in OS or PFS (P = .99 and 0.96, respectively) (Figure 1). None of the 4 patients who met strict Baltimore criteria were alive at 1 year (supplemental Figure 1). Patients with anicteric VOD/SOS had similar OS and PFS compared with patients with icteric VOD/SOS (P = .87 and 0.79) (supplemental Figure 2). Among the 6 patients with very severe VOD/SOS in our cohort who had pulmonary dysfunction at diagnosis, only 2 died before day 100.
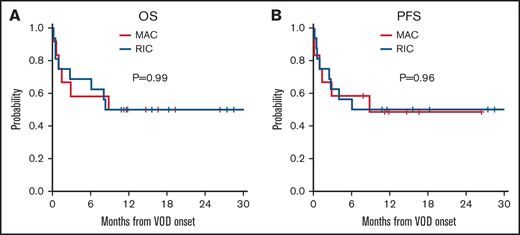
Kaplan-Meier curves based on patients who received MAC and RIC regimens. (A) OS. (B) PFS.
Kaplan-Meier curves based on patients who received MAC and RIC regimens. (A) OS. (B) PFS.
Factors associated with response to defibrotide
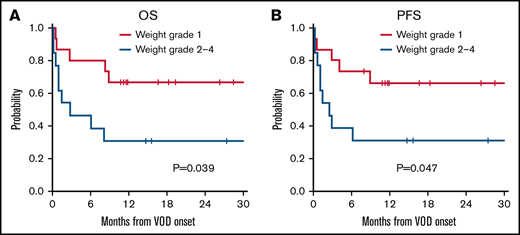
No predictors were significantly associated with response. However, patients who experienced grade II to IV weight gain, defined as a weight gain >5% from pretransplant to VOD/SOS diagnosis, were associated with a significantly poorer OS and PFS compared with a grade I weight increase (18-month OS, 67% vs 31% for OS, respectively [P = .039]; 18-month PFS, 66% vs 31% for PFS [P = .047]) (Figure 2). Grade severity, graft source, type of conditioning regimen, and pulmonary dysfunction at diagnosis did not significantly affect response to defibrotide (P = .15, 0.14, 1, and 0.62).
Adverse events after defibrotide therapy
All patients who received defibrotide experienced at least 1 adverse event (Table 3). Hematuria occurred in 43% (n = 12). Grade III hypotension, defined as requiring ≥1 pressor drug, occurred in 11% (n = 3). Epistaxis occurred in 18% (n = 5) of patients. Grade III/IV hemorrhagic adverse events were rare and included 1 patient with pulmonary hemorrhage and 1 patient with upper gastrointestinal hemorrhage. Both of these patients had very severe VOD/SOS in addition to a weight gain >10%, with progressive VOD/SOS at the time of each event. Therapy was interrupted for minor surgical procedures (n = 8), platelet count <30 000 to 50 000/µL (n = 5), fibrinogen <150 mg/dL (n = 1), pulmonary hemorrhage (n = 1), and grade III to IV gastrointestinal hemorrhage (n = 1). Six patients (21%) experienced relapse of their underlying hematologic malignancy after defibrotide therapy; however, in each case, the relapse was not considered related by their treating physicians and was due to their underlying high-risk disease.
Treatment-related adverse events
| Adverse event | All HSCT (N = 28) | Mild-moderate-severe VOD/SOS (n = 11) | Very severe VOD/SOS (n = 17) |
|---|---|---|---|
| Hypotension, n (%) | 3 (11) | 0 | 3 (18) |
| Lower gastrointestinal hemorrhage, n (%) | 1 (4) | 0 | 1 (6) |
| Epistaxis, n (%) | 5 (18) | 1 (9) | 4 (24) |
| Grade III/IV pulmonary hemorrhage, n (%) | 1 (4) | 0 | 1 (6) |
| Grade III/IV upper gastrointestinal hemorrhage, n (%) | 1 (4) | 0 | 1 (6) |
| Hematuria, n (%) | 12 (43) | 3 (27) | 9 (53) |
| Adverse event | All HSCT (N = 28) | Mild-moderate-severe VOD/SOS (n = 11) | Very severe VOD/SOS (n = 17) |
|---|---|---|---|
| Hypotension, n (%) | 3 (11) | 0 | 3 (18) |
| Lower gastrointestinal hemorrhage, n (%) | 1 (4) | 0 | 1 (6) |
| Epistaxis, n (%) | 5 (18) | 1 (9) | 4 (24) |
| Grade III/IV pulmonary hemorrhage, n (%) | 1 (4) | 0 | 1 (6) |
| Grade III/IV upper gastrointestinal hemorrhage, n (%) | 1 (4) | 0 | 1 (6) |
| Hematuria, n (%) | 12 (43) | 3 (27) | 9 (53) |
Discussion
VOD/SOS is a well-recognized hepatic complication after HSCT for which defibrotide is the only FDA-approved therapy to date. In the phase 3 trial,8 defibrotide was associated with a 23% propensity-adjusted improvement in day 100 survival for patients with severe VOD/SOS compared with matched historical control subjects. Although there have been many publications regarding the use of defibrotide from clinical trials and treatment investigational new drug application studies, there have been no reports on the real-world postmarketing use of defibrotide after its FDA approval in 2016.12,15
The current study found a VOD/SOS complete resolution rate of 75% and a very encouraging OS of 64% at day 100. One-year treatment-related mortality was 39%. Even in the group with very severe VOD/SOS, day 100 survival was 53%, which compares favorably to the 38.2% day 100 OS reported in the phase 3 study that led to defibrotide’s FDA approval. This could potentially be due to differences in VOD/SOS severity definitions between the EBMT severity criteria and the Baltimore criteria used in the phase 3 trial, our center’s experience with defibrotide, or other variables. Compared with the trial population, the VOD/SOS population in our center had anicteric VOD, later onset, and higher propensity for acute renal dysfunction. This may reflect the fact that our VOD/SOS cases are more associated with the use of sirolimus with tacrolimus as GVHD prophylaxis, and the routine use of ursodeoxycholic acid in the first 100 days after HSCT, which could lower the serum total bilirubin in these study patients with VOD/SOS.
As a center that commonly uses tacrolimus with sirolimus, especially in our RIC transplant cases, we have observed a higher incidence of VOD/SOS; this is particularly true for anicteric VOD/SOS that presents with early renal dysfunction, portal venous flow abnormalities, and a delayed or minimal rise in total bilirubin. In these anicteric cases, the Baltimore criteria would not be able to establish the diagnosis or not until late in the disease course.
The encouraging results after defibrotide therapy at our center may not be applicable to other real-world settings because transplant center practices and experiences with VOD/SOS can vary widely. As a center that has a long history of treating VOD/SOS and pioneering early defibrotide trials, our clinicians may be more in-tuned to this diagnosis, and it is possible that patients with milder disease may have been treated at our center. However, it is noteworthy that even though many of our cases are anicteric VOD/SOS, most still had organ dysfunction (renal [75%] and lung [21%]), as well ascites (93%) and hepatomegaly (61%), indicating some high-risk features.
We found that response rates and survival after defibrotide were similar after MAC and RIC transplants, as well as in icteric vs anicteric VOD/SOS. The only factor in our cohort that predicted poor response was degree of weight gain (>5%). The paucity of patients in this group limits our ability to draw firm conclusions, but this finding seems consistent with the Bearman criteria for VOD/SOS severity in which the degree of weight gain is one of the key predictors of survival.16
Defibrotide was generally well tolerated in our study cohort, and the adverse events we observed seemed comparable to those reported in previous clinical trials.17 Duration of therapy varied slightly from the 21-day FDA-approved recommendation, but this finding is reflective of the real-world experience in which physicians may be tailoring therapy and duration according to patient response. Despite the expected thrombocytopenia and coagulopathy associated with VOD/SOS, and the fact that defibrotide has antithrombotic properties, bleeding complications were manageable and mainly restricted to mild hematuria and epistaxis, and there were only 2 cases of severe hemorrhage. Defibrotide therapy was interrupted in 17 patients; 5 of these patients had severe thrombocytopenia and 1 patient had hypofibrinogenemia, with the majority of interruptions, due to lower laboratory values, occurring before 2019. Importantly, with continued experience, clinicians at our institution have in recent years pursued strategies to minimize dose interruptions to maximize treatment response while maintaining vigilance regarding any adverse effects. These strategies include not holding >1 dose of defibrotide for minor procedures such as line placement or paracentesis and not interrupting dosing due to thrombocytopenia or elevated prothrombin time/international normalized ratio in the absence of clinical bleeding.
The current study had some limitations owing to its single-center retrospective design, and its small sample size precludes more definitive statistical analyses. In addition, our encouraging results may not extend to other transplant centers. The retrospective nature of our study could also have led to some underassessment of adverse events because these were retrospectively identified based on laboratory and chart review. Nonetheless, our series of 28 patients represents one of the largest single-center VOD/SOS series treated in recent years and the first to describe the real-world experience for defibrotide in the management of VOD/SOS since FDA approval. Our series is also among the first to report on the encouraging outcomes of defibrotide treatment in anicteric VOD and VOD/SOS after RIC transplant.
In conclusion, this real-world study found that defibrotide as treatment of VOD/SOS in adults after allogeneic HSCT is associated with a 75% complete response rate and encouraging day 100 survival, with relatively few severe adverse events. Our results suggest that early diagnosis, prompt initiation of defibrotide, and minimization of dosing interruptions may be key to successful treatment of this very challenging HSCT complication.
Authorship
Contribution: M.N. was involved in the conception and design of the study, acquisition of data, analysis and interpretation of data, and writing of the manuscript; V.T.H., P.G.R., and R.R. were involved in design of the study and writing of the manuscript; and H.T.K. was involved in the analysis and interpretation of data. All authors contributed to the development of the manuscript and revision and provided their final approval of the manuscript.
Conflict-of-interest disclosure: P.G.R. has served on advisory committees for Jazz Pharmaceuticals. C.C. has served as an educational consultant to Jazz Pharmaceuticals. J.H.A. has served on the scientific advisory board for Merck, Snipr Biome, and Pharmacosmos; and served on the data and safety monitoring board for Momenta and CSL Behring. V.T.H. has received research funding support from Jazz Pharmaceuticals; and served as a consultant for Alexion, Janssen, and Omeros. The remaining authors declare no competing financial interests.
Correspondence: Vincent T. Ho, 450 Brookline Ave, Boston, MA 02215; e-mail: Vincent_ho@dfci.harvard.edu.

